- Gaucher's disease
-
Gaucher's disease Classification and external resources 
Acid beta-glucosidaseICD-10 E75.2 (ILDS E75.220) ICD-9 272.7 OMIM 230800 230900 231000 DiseasesDB 5124 MedlinePlus 000564 eMedicine ped/837 derm/709 MeSH D005776 GeneReviews Gaucher Disease Gaucher's disease (
 /ɡoʊˈʃeɪz/) is a genetic disease in which a fatty substance (lipid) accumulates in cells and certain organs. Gaucher's disease is the most common of the lysosomal storage diseases.[1]:536 It is caused by a hereditary deficiency of the enzyme glucosylceramidase. The enzyme acts on the fatty acid glucosylceramide. When the enzyme is defective, glucosylceramide accumulates, particularly in white blood cells, most often macrophages (mononuclear leukocytes). glucosylceramidase can collect in the spleen, liver, kidneys, lungs, brain and bone marrow.
/ɡoʊˈʃeɪz/) is a genetic disease in which a fatty substance (lipid) accumulates in cells and certain organs. Gaucher's disease is the most common of the lysosomal storage diseases.[1]:536 It is caused by a hereditary deficiency of the enzyme glucosylceramidase. The enzyme acts on the fatty acid glucosylceramide. When the enzyme is defective, glucosylceramide accumulates, particularly in white blood cells, most often macrophages (mononuclear leukocytes). glucosylceramidase can collect in the spleen, liver, kidneys, lungs, brain and bone marrow.Symptoms may include enlarged spleen and liver, liver malfunction, skeletal disorders and bone lesions that may be painful, severe neurologic complications, swelling of lymph nodes and (occasionally) adjacent joints, distended abdomen, a brownish tint to the skin, anemia, low blood platelets and yellow fatty deposits on the white of the eye (sclera). Persons affected most seriously may also be more susceptible to infection. Some forms of Gaucher's disease may be treated with enzyme replacement therapy.
The disease is caused by a recessive mutation in a gene located on chromosome 1 and affects both males and females. About 1 in 100 people in the United States are carriers of the most common type of Gaucher disease, while the carrier rate among Ashkenazi Jews is 8.9% while the birth incidence is 1 in 450.[2]
The disease is named after the French doctor Philippe Gaucher, who originally described it in 1882.[3]
Contents
Classification
Gaucher's disease has three common clinical subtypes.
- Type I (or non-neuropathic type) is the most common form of the disease, occurring in approximately 1 in 50,000 live births. It occurs most often among persons of Ashkenazi Jewish heritage. Symptoms may begin early in life or in adulthood and include enlarged liver and grossly enlarged spleen (together hepatosplenomegaly); the spleen can rupture and cause additional complications. Skeletal weakness and bone disease may be extensive. Spleen enlargement and bone marrow replacement cause anemia, thrombocytopenia and leukopenia. The brain is not affected pathologically, but there may be lung and, rarely, kidney impairment. Patients in this group usually bruise easily (due to low levels of platelets) and experience fatigue due to low numbers of red blood cells. Depending on disease onset and severity, type 1 patients may live well into adulthood. Many patients have a mild form of the disease or may not show any symptoms.
- Type II (or acute infantile neuropathic Gaucher's disease) typically begins within 6 months of birth and has an incidence rate of approximately 1 in 100,000 live births. Symptoms include an enlarged liver and spleen, extensive and progressive brain damage, eye movement disorders, spasticity, seizures, limb rigidity, and a poor ability to suck and swallow. Affected children usually die by age 2.
- Type III (the chronic neuropathic form) can begin at any time in childhood or even in adulthood, and occurs in approximately 1 in 100,000 live births. It is characterized by slowly progressive but milder neurologic symptoms compared to the acute or type 2 version. Major symptoms include an enlarged spleen and/or liver, seizures, poor coordination, skeletal irregularities, eye movement disorders, blood disorders including anemia and respiratory problems. Patients often live into their early teen years and adulthood.
These subtypes have come under some criticism for not taking account of the full spectrum of observable symptoms (the phenotypes.[1]) There are also compound heterozygous variations which considerably increase the complexity of predicting disease course.
Signs and symptoms
- Painless hepatomegaly and splenomegaly: the size of the spleen can be 1500-3000 ml, as opposed to the normal size of 50-200 ml. Splenomegaly may decrease the affected individual's capacity for eating by exerting pressure on the stomach. While painless, enlargement of spleen increases the risk of splenic rupture.
- Hypersplenism and pancytopenia: the rapid and premature destruction of blood cells, leading to anemia, neutropenia, leukopenia, and thrombocytopenia (with an increased risk of infection and bleeding).
- Cirrhosis of the liver is rare
- Severe pain associated with joints and bones, frequently presenting in hips and knees.
- Neurological symptoms occur only in some types of Gaucher's (see below):
- Type II: serious convulsions, hypertonia, mental retardation, apnea.
- Type III: muscle twitches known as myoclonus, convulsions, dementia, ocular muscle apraxia.
- Osteoporosis: 75% develop visible bony abnormalities due to the accumulated glucosylceramide. A deformity of the distal femur in the shape of an Erlenmeyer flask is commonly described (aseptic necrosis of the femur joint).
- Yellowish-brown skin pigmentation
Pathophysiology
The disease is caused by a defect in housekeeping gene lysosomal gluco-cerebrosidase (also known as beta-glucosidase, EC 3.2.1.45, PDB 1OGS) on the first chromosome (1q21). The enzyme is a 55.6 KD, 497 amino acids long protein that catalyses the breakdown of glucosylceramidase, a cell membrane constituent of red and white blood cells. The macrophages that clear these cells are unable to eliminate the waste product, which accumulates in fibrils, and turn into Gaucher cells, which appear on light microscopy to resemble crumpled-up paper.
In the brain (type II and III), glucosylceramidase accumulates due to the turnover of complex lipids during brain development and the formation of the myelin sheath of nerves.
Different mutations in the beta-glucosidase determine the remaining activity of the enzyme, and, to a large extent, the phenotype.
Heterozygotes for particular acid beta-glucosidase mutations carry about a fivefold risk of developing Parkinson's disease, making this the most common known genetic risk-factor for Parkinson's.[4][5] A study of 1525 Gaucher patients in the United States suggested that while cancer risk is not elevated, particular malignancies (non-Hodgkin lymphoma, melanoma and pancreatic cancer) occurred at a 2-3 times higher rate.[6]
Genetics
The three types of Gaucher's disease are inherited in an autosomal recessive fashion. Both parents must be carriers in order for a child to be affected. If both parents are carriers, there is a one in four, or 25%, chance with each pregnancy for an affected child. Genetic counseling and genetic testing is recommended for families who may be carriers of mutations.
Each type has been linked to particular mutations. In all, there are about 80 known mutations, grouped into three main types:[7]
- Type I (N370S homozygote), the most common, also called the "non-neuropathic" type occurs mainly in Ashkenazi Jews, at 100 times the occurrence in the general populace. The median age at diagnosis is 28 years of age,[8] and life expectancy is mildly decreased.[9] There are no neurological symptoms.
- Type II (1 or 2 alleles L444P) is characterized by neurological problems in small children. The enzyme is hardly released into the lysosomes. Prognosis is dismal: most die before reaching the third birthday.
- Type III (also 1-2 copies of L444P, possibly delayed by protective polymorphisms) occurs in Swedish patients from the Norrbotten region. This group develops the disease somewhat later, but most die before their 30th birthday.
Diaz et al. suggest that the Gaucher-causing mutations entered the Ashkenazi Jewish gene pool in the early Middle Ages (48-55 generations ago).[10]
Diagnosis
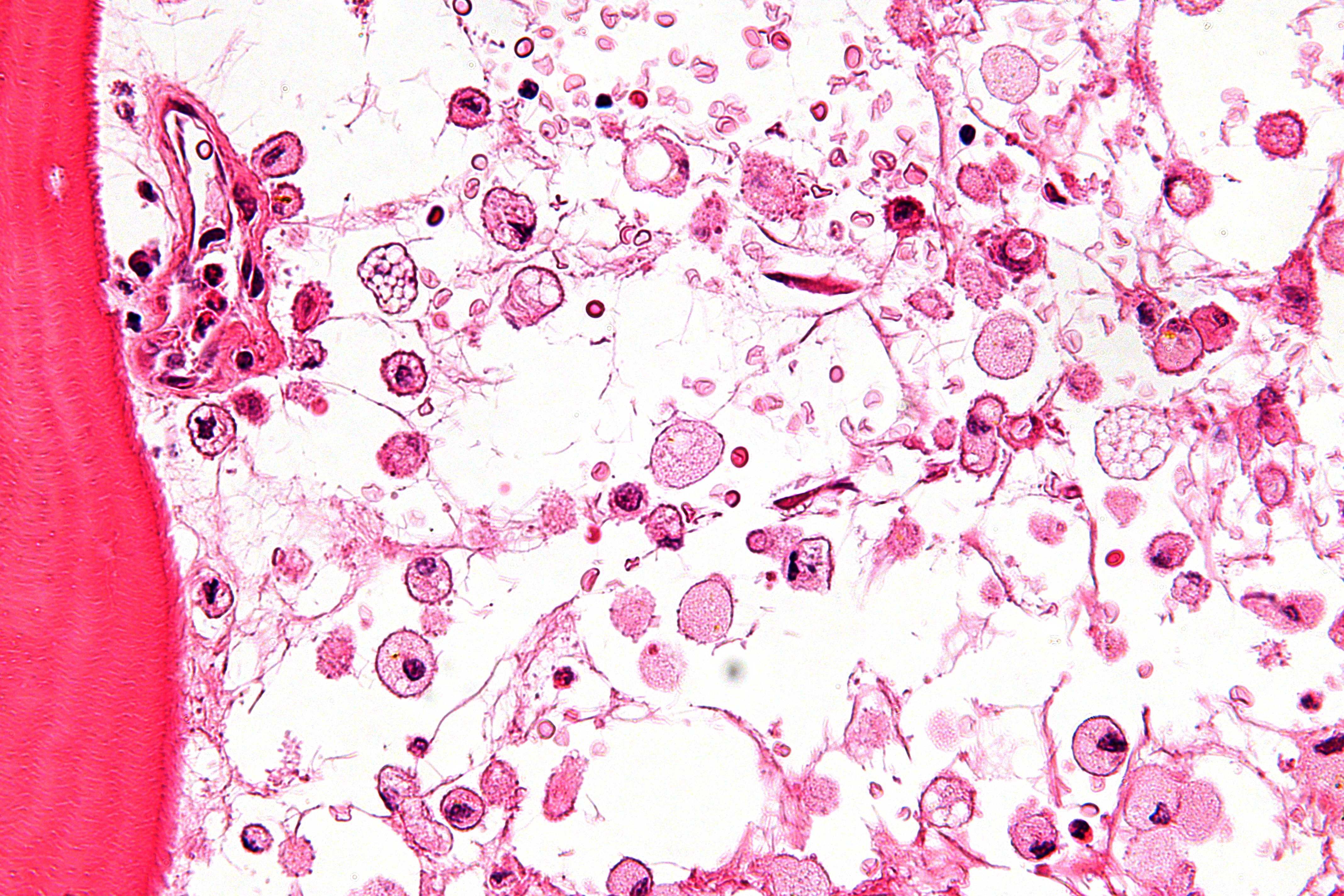
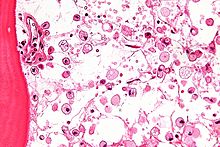 Micrograph showing crinkled paper macrophages in the marrow space in a case of Gaucher disease. H&E stain.
Micrograph showing crinkled paper macrophages in the marrow space in a case of Gaucher disease. H&E stain.
A definitive diagnosis is made with genetic testing. As there are numerous different mutations, sequencing of the beta-glucosidase gene is sometimes necessary to confirm the diagnosis. Prenatal diagnosis is available, and is useful when there is a known genetic risk factor.
A diagnosis can also be implied by biochemical abnormalities such as high alkaline phosphatase, angiotensin-converting enzyme (ACE) and immunoglobulin levels, or by cell analysis showing "crinkled paper" cytoplasm and glycolipid-laden macrophages.
Some lysosomal enzymes are elevated, including tartrate-resistant acid phosphatase, hexosaminidase, and a human chitinase, chitotriosidase. This latter enzyme has proved to be very useful for monitoring Gaucher's disease activity in response to treatment, and may reflect the severity of the disease
Treatment
For type 1 and most type 3 patients, enzyme replacement treatment with intravenous recombinant glucocerebrosidase (imiglucerase) can dramatically decrease liver and spleen size, reduce skeletal abnormalities, and reverse other manifestations. This treatment costs approximately $200,000 annually for a single patient and should be continued for life. The rarity of the disease means that dose-finding studies have been difficult to conduct, so there remains controversy over the optimal dose and dosing frequency.[8] Due to the low incidence, this has become an orphan drug in many countries, meaning that a government recognizes and accommodates the financial constraints that limit research into drugs that address a small population.Velaglucerase alfa was approved by the Food and Drug Administration (FDA) as an alternative treatment on February 26, 2010.[11]
Successful bone marrow transplantation cures the non-neurological manifestations of the disease, because it introduces a monocyte population with active beta-glucosidase. However, this procedure carries significant risk and is rarely performed in Gaucher patients. Surgery to remove the spleen (splenectomy) may be required on rare occasions if the patient is anemic or when the enlarged organ affects the patient’s comfort. Blood transfusion may benefit some anemic patients. Other patients may require joint replacement surgery to improve mobility and quality of life. Other treatment options include antibiotics for infections, antiepileptics for seizures, bisphosphonates for bone lesions, and liver transplants. Substrate reduction therapy may prove to be effective in stopping Type 2, as it can cross through the blood barrier into the brain. There is currently no effective treatment for the severe brain damage that may occur in patients with types 2 and 3 Gaucher disease. Gene therapy may be a future step.
Gaucher's disease has recently become a target for more than one effort at pharmacological chaperoning, which involves the use of orally administered drugs that operate at a molecular level. Miglustat is one of these oral drugs. It was approved for the treatment of this disease in 2003. As of June 2009[update], another oral drug, isofagomine tartrate, is under development.
Epidemiology
The National Gaucher Foundation states that around 1 in 100 people in the general U.S. population is a carrier for type 1 Gaucher's disease, giving a prevalence of 1 in 40,000: among Ashkenazi Jews the rate of carriers is considerably higher, at roughly 1 in 15.[12]
Type 2 Gaucher's disease shows no particular preference for any ethnic group.
Type 3 Gaucher's disease is especially common in the population of the Northern Swedish region of Norrbotten where the incidence of the disease is 1 in 50,000.
History
The disease was first recognised by the French doctor Philippe Gaucher, who originally described it in 1882 and lent his name to the condition.[3] The biochemical basis for the disease would be elucidated in 1965.[13] The first effective treatment for the disease, the drug Ceredase, was approved by the FDA in April 1991. An improved drug, Cerezyme, was approved by the FDA in May 1994 and has replaced the use of Ceredase.
Additional images
-

Sphingolipidoses
See also
- Niemann-Pick disease
- Fabry disease
- Tay-Sachs
- Krabbe disease
- Metachromatic leukodystrophy
- Medical genetics of Ashkenazi Jews
References
- ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ^ Zimran A, Gelbart T, Westwood B, Grabowski GA, Beutler E (1991). "High frequency of the Gaucher disease mutation at nucleotide 1226 among Ashkenazi Jews". Am. J. Hum. Genet. 49 (4): 855–859. PMC 1683177. PMID 1897529. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1683177.
- ^ a b Gaucher PCE (1882). De l'epithelioma primitif de la rate, hypertrophie idiopathique de la rate sans leucemie [academic thesis]. Paris, France.
- ^ Jacquelyn K Beals (November 19, 2008). "ASHG 2008: Gaucher Disease Mutation Carriers at Higher Risk for Parkinson's Disease". Medscape Medical News.
- ^ Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R (2004). "Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews". N. Engl. J. Med. 351 (19): 1972–7. doi:10.1056/NEJMoa033277. PMID 15525722.
- ^ Landgren O, Turesson I, Gridley G, Caporaso NE (2007). "Risk of Malignant Disease Among 1525 Adult Male US Veterans With Gaucher Disease". Archives of Internal Medicine 167 (11): 1189–1194. doi:10.1001/archinte.167.11.1189. PMID 17563029.
- ^ Online 'Mendelian Inheritance in Man' (OMIM) 606463
- ^ a b Grabowski GA (2008). "Phenotype, diagnosis, and treatment of Gaucher's disease". Lancet 372 (9645): 1263–1271. doi:10.1016/S0140-6736(08)61522-6.
- ^ Weinreb NJ, Deegan P, Kacena KA, et al. (December 2008). "Life expectancy in Gaucher disease type 1". Am. J. Hematol. 83 (12): 896–900. doi:10.1002/ajh.21305. PMID 18980271.
- ^ Diaz GA, Gelb BD, Risch N, et al. (2000). "Gaucher disease: the origins of the Ashkenazi Jewish N370S and 84GG acid beta-glucosidase mutations". Am. J. Hum. Genet. 66 (6): 1821–32. doi:10.1086/302946. PMC 1378046. PMID 10777718. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1378046.
- ^ http://www.medicalnewstoday.com/articles/180630.php
- ^ "National Gaucher Foundation". http://www.gaucherdisease.org/prevalence.php. Retrieved 2007-05-30.
- ^ Brady RO, Kanfer JN, Shapiro D (1965). "Metabolism of glucosylceramidase. II. Evidence of an enzymatic deficiency in Gaucher's disease". Biochem. Biophys. Res. Commun. 18 (2): 221–5. doi:10.1016/0006-291X(65)90743-6. PMID 14282020.
External links
Listen to this article (info/dl)
To sphingosineNCL Other Cerebrotendineous xanthomatosis · Cholesteryl ester storage disease (Lysosomal acid lipase deficiency/Wolman disease) · Sea-blue histiocyte syndromeCategories:- Autosomal recessive disorders
- Rare diseases
- Lipid storage disorders
- Ashkenazi Jews topics
- Skin conditions resulting from errors in metabolism
Wikimedia Foundation. 2010.
Look at other dictionaries:
Gaucher's disease — Gau·cher s disease .gō shāz n a rare hereditary disorder of lipid metabolism that is caused by an enzyme deficiency of glucocerebrosidase, that is characterized by enormous enlargement of the spleen, pigmentation of the skin, and bone lesions,… … Medical dictionary
Gaucher cells disease — Gau·cher cells, disease (go shaґ) [Phillippe Charles Ernest Gaucher, French physician, 1854–1918] see under cell and disease … Medical dictionary
Gaucher's disease — a genetically determined (autosomal recessive) disease resulting from the deposition of glucocerebrosides (see cerebroside) in the brain and other tissues (especially bone). It causes mental retardation, abnormal limb posture and spasticity, and… … The new mediacal dictionary
Gaucher's disease — noun Etymology: Philippe C. E. Gaucher died 1918 French physician Date: 1902 a rare hereditary disorder of lipid metabolism caused by an enzyme deficiency and characterized by enlargement of the spleen and liver, bone lesions, and neurological… … New Collegiate Dictionary
Gaucher's disease — /goh shayz /, Pathol. a rare inherited disorder of fat metabolism that causes spleen and liver enlargement, abnormal fragility and pain of the bones, and progressive neurologic disturbances, leading to early death. [after Philippe C. Ernest… … Universalium
Gaucher's disease — Familial autosomal recessive defect of glucocerebrosidase ( b glucosidase), most common in Ashkenazi Jews. Associated with hepatosplenomegaly (enlargement of liver and spleen) and, in severe early onset forms of the disease, with neurological… … Dictionary of molecular biology
Gaucher's disease — noun A genetic disease in which lipid accumulates in cells and certain organs … Wiktionary
Gaucher's disease — genetic disease that causes an accumulation of fats in storage cells as a result of a lack of enzyme activity in breaking down molecules composed of sugar and fats … English contemporary dictionary
Gaucher's disease — noun a rare chronic disorder of lipid metabolism of genetic origin • Hypernyms: ↑lipidosis, ↑monogenic disorder, ↑monogenic disease … Useful english dictionary
Gaucher disease — ▪ disease rare inherited metabolic disorder characterized by anemia, mental and neurologic impairment, yellowish pigmentation of the skin, enlargement of the spleen, and bone deterioration resulting in pathological fractures. Gaucher… … Universalium
 18+© Academic, 2000-2024
18+© Academic, 2000-2024- Contact us: Technical Support, Advertising
Dictionaries export, created on PHP, Joomla, Drupal, WordPress, MODx.