- Molecular Inversion Probe
-
Molecular Inversion Probe (MIP)[1] belongs to the class of Capture by Circularization molecular techniques [1] for performing genomic partitioning, a process through which one captures and enriches specific regions of the genome[2]. Probes used in this technique are single stranded DNA molecules and, similar to other genomic partitioning techniques, contain sequences that are complementary to the target in the genome; these probes hybridize to and capture the genomic target. MIP stands unique from other genomic partitioning strategies in that MIP probes share the common design of two genomic target complementary segments separated by a linker region. With this design, when the probe hybridizes to the target, it undergoes an inversion in configuration (as suggested by the name of the technique) and circularizes. Specifically, the two target complementary regions at the 5’ and 3’ ends of the probe become adjacent to one another while the internal linker region forms a free hanging loop. The technology has been used extensively in the HapMap project for large-scale SNP genotyping[3] as well as for studying gene copy alterations[4] and characteristics of specific genomic loci[2][5] to identify biomarkers for different diseases such as cancer. Key strengths of the MIP technology include its high specificity to the target and its scalability for high-throughput, multiplexed analyses where tens of thousands of genomic loci are assayed simultaneously.
Contents
Technique Procedure
 Features of a typical Molecular Inversion Probe
Features of a typical Molecular Inversion Probe
 Procedure for genomic regions capture using Molecular Inversion Probes
Procedure for genomic regions capture using Molecular Inversion ProbesMolecular Inversion Probe Structure
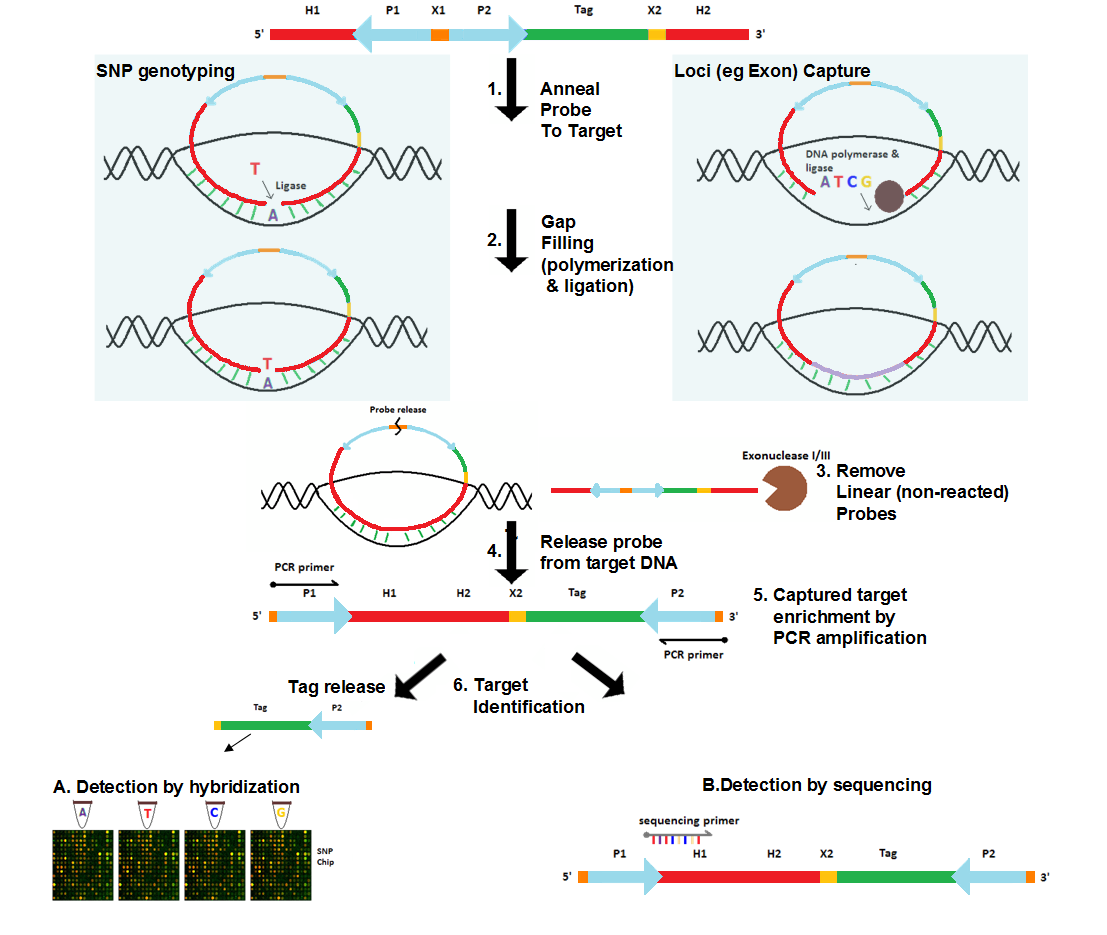
The probes are designed with sequences that are complementary to the genomic target at its 5’ and 3’ ends [2][3][6]. The internal region contains two universal PCR primer sites that are common to all MIPs as well as a probe-release site , which is usually a restriction site [3]. If the identification of the captured genomic target is performed using array-based hybridization approaches, the internal region may optionally contain a probe-specific tag sequence that uniquely identifies the given probe as well as a tag-release site, which, similar to the probe-release site, is also a restriction site.
Protocol
- Anneal probe to genomic target DNA
Probes are added to the genomic DNA sample. After a denaturation followed by an annealing step, the target-complementary ends of the probe are hybridized to the target DNA. The probes then undergo circularization in this process. These probes, however, are designed such that a gap delimited by the hybridized ends of the probes remains over the target region. The size of the gap ranges from a single nucleotide for SNP genotyping [3] to several hundred nucleotides for loci capture (e.g. exome capture) [5].
- Gap filling
The gap is filled by DNA polymerase using free nucleotides and the ends of the probe are ligated by ligase, resulting in a fully circularized probe.
- Remove non-reacted probes
Since gap filling is not performed for non-reacted probes, they remain linear. Exonuclease treatment removes these non-reacted probes as well as any remaining linear DNA in the reaction.
- Probe release
In some versions of the protocol, the probe-release site (commonly a restriction site) is cleaved by restriction enzymes such that the probe becomes linearized. In this linearized probe the universal PCR primer sequences are located at the 5’ and 3’ ends and the captured genomic target becomes part of the internal segment of the probe. Other protocols leave the probe as a circularized molecule.
- Captured target enrichment
If the probe is linearized, traditional PCR amplification is performed to enrich the captured target using the universal primers of the probe. Otherwise, rolling circle amplification is performed for the circular probe.
- Captured target identification
The captured target can be identified either via array-based hybridization approaches [3] or by sequencing of the target [5]. If array-based approach is used, the probe may optionally contain a probe-specific tag that uniquely identifies the probe as well as the genomic region targeted by it. The tags from each probe are released by cleaving the tag release site with restriction enzymes. These tags are then hybridized to the sequences that are placed on the array and are complementary to them. The captured target can also be identified by sequencing the probe, now also containing the target. Traditional Sanger sequencing or cheaper, more high-throughput technologies such as SOLiD, Illumina or Roche 454 can be used for this purpose.
Multiplex analysis
Although each probe examines one specific genomic locus, multiple probes can be combined into a single tube for multiplexed assay that simultaneously examines multiple loci. Currently, multiplexed MIP analysis can examine more than 55,000 loci in a single assay [2].Technique Development History
 Schematics of Padlock Probes, Molecular Inversion Probes and Connector Inversion Probes
Schematics of Padlock Probes, Molecular Inversion Probes and Connector Inversion ProbesPadlock Probe
The design of the molecular inversion probes (MIP) originated from padlock probes, a molecular biology technique first reported by Nilsson et al. in 1994 [7]. Similar to MIP, padlock probes are single stranded DNA molecules with two 20-nucleotide long segments complementary to the target connected by a 40-nucleotide long linker sequence. When the target complementary regions are hybridized to the DNA target, the padlock probes also become circularized. However, unlike MIP, padlock probes are designed such that the target complementary regions span the entire target region upon hybridization, leaving no gaps. Thus, padlock probes are only useful for detecting DNA molecules with known sequences.
Nilsson et al.[7] demonstrated the use of padlock probes to detect numerous DNA targets, including a synthetic oligonucleotide and a circular genomic clone. Padlock probes have high specificity towards their target and can distinguish target molecules that closely resemble one another. Nilsson et al.[7] also demonstrated the use of padlock probes to differentiate between a normal and a mutant cystic fibrosis conductance receptor (CFCR) where the CFCR mutant had a 3bp deletion corresponding to one of the ends of the probe. Since ligation requires the ends of the probe to be immediately adjacent to one another when hybridized to the target, the 3bp deletion in the mutant prevented successful ligation. Padlock probes were also successfully used for in situ hybridization to detect alphoid repeats specific to chromosome 12 in a sample of chromosomes in metastasis state. Here, traditional, linear oligonucleotide probes failed to yield results [7]. Thus, padlock probes possess sufficient specificity to detect single copy elements in the genome [7].
Molecular Inversion Probe
In order to perform SNP genotyping, Hardenbol et al.[3] modified padlock probes such that when the probe is hybridized to the genomic target, there is a gap at the SNP position. Gap filling using a nucleotide that is complementary to the nucleotide at the SNP location determines the identity of the polymorphism. This design brings numerous benefits over the more traditional padlock probe technique. Using multiple padlock probes specific to a plausible SNP requires careful balancing of the concentration of these allele specific probes to ensure SNP counts at a given locus are properly normalized [3]. In addition, with this design, bad probes affect all genotypes at a given locus equally [3]. For instance, since MIP probes can assay multiple genotypes at a particular genomic locus, if the probe for a given locus does not work (e.g. fails to properly hybridize to the genomic target), none of the genotypes at this locus will be detected. In contrast, for padlock probes, one needs to design a distinct padlock probe to detect each plausible genotype a given locus (e.g. one padlock probe is needed for detecting "A" at a given SNP locus and another padlock probe is needed for detecting "T" at the locus). Thus, a bad padlock probe will only affect the detection of the specific genotype that the probe is designed to detect whereas a bad MIP probe will affect all genotypes at the locus. Using MIP, one avoids potential incorrect SNP calling since if the probe designed to assay a given locus does not work, no data is generated for this locus and no SNP calling is performed.
In their procedure, Hardenbol et al.[3] assayed more than 1000 SNP loci simultaneously in a single tube where the tube contained more than 1000 probes with distinct designs. The pool of probes was aliquoted into four tubes for four different reactions. In each reaction, a distinct nucleotide (A, T, C or G) was used for gap filling. Only when the nucleotide at the SNP locus was complementary to the applied nucleotide would the gap be closed by ligation and the probe be circularized. Identification of the captured SNPs was performed on genotyping arrays where each spot on the array contained sequences complementary to the locus-specific tags in the probes. Since the DNA array costs is a major contributor to the cost of this technique, the performance of four-chip-one-color detection was compared to two-chip-two color detection. The results were found to be similar in terms of SNP call rate and signal-to-noise ratio [3].
In a recent report[6], this group successfully increased the level of multiplexing to simultaneously assay more than 10,000 SNP loci, using 12,000 distinct probes. The study examined SNP polymorphisms in 30 trio samples (each trio consisted of a mother, father and their child). Knowing the genotypes of the parents, the accuracy of the SNP genotypes predicted in the child was determined by examining whether a concordance existed between the expected Mendelian inheritance patterns and the predicted genotypes. Trio concordance rate has been found to be > 99.6%[1]. In addition, a set of MIP-specific performance metrics was developed. This work set the framework for high-throughput SNP genotyping in the HapMap project [6].
Connector Inversion Probe
To capture longer genomic regions than a single nucleotide, Akhras et al.[5] modified the design of MIP by extending the gap delimited by the hybridized probe ends and named the design Connector Inversion Probe (CIP). The gap corresponds to the genomic region of interest to be captured (e.g. exons). Gap filling reaction is achieved with DNA polymerase, using all four nucleotides. Identification of the captured regions can then be done by sequencing them using locus-specific primers that map to one of the target complementary ends of the probes.
Akhras et al.[5] also developed the multiplexing multiplex padlocks (MMP) barcode system in order to lower the costs of reagents. A single assay might involve DNA samples from multiple individuals and examine multiple genomic loci in each individual. A DNA barcode system that uniquely identifies each plausible combination of individual and genomic locus is represented as DNA tags that were inserted into the linker region of the probes. Thus, sequences from the captured regions would include the barcode, allowing the non-ambiguous determination of the individual and the genomic locus that the captured region belongs to.
This group has also developed a software for designing locus-specific CIPs (CIP creator 1.0.1).
Application
Molecular Inversion Probe (MIP) is one of the techniques widely used to capture a small region of the genome for further examination. With the invention of the next generation sequencing technologies, the cost of sequencing whole genomes has decreased dramatically, however the cost is still too high for these sequencing machines to be used in practice in every laboratory. Instead, different genome partitioning techniques can be used to isolate smaller but highly specific regions of the genome for further analysis. MIP, for instance, can be used to capture targets for SNPgenotyping, copy number variation or allelic imbalance studies, to name a few.
SNP Genotyping
In SNP genotyping, the probes are separated into four reactions and a different type of nucleotide is added to each reaction. If the SNP at the target region is complementary to the added nucleotide, the ligation is successful and the probe becomes fully circularized. Since each probe hybridizes to exactly one SNP target in the genome, successfully circularized probes provide the nucleotide identities of the SNPs. The tag sequences from the four nucleotide-specific reactions are then hybridized to either four genotyping arrays or two, dual-colour arrays (one channel for each reaction). Analyzing which spots on the array are bound by the tags allows the determination of the SNP identities at the genomic loci represented by those tags.
The SNPs targeted by MIP can then be used in areas of research such as quantitative trait loci (QTL) analysis or genome-wide association studies (GWAS) where the SNPs are used in either indirect linkage disequilibrium studies or directly screened for causative mutations.
Copy Number Variation Detection
Molecular inversion probe technique can also be used for copy number variation (CNV) detection. This dual role in SNP genotyping as well as CNV analysis of MIP is similar to the high-density SNP genotyping arrays which have recently been used for CNV detection and analysis as well. These techniques extract the allele-specific signal intensities from genotyping data and use that to generate CNV results. These techniques have higher precision and resolution than traditional techniques such as G-banded karyotypic analyses, fluorescence in situ hybridization (FISH) or array comparative genomic hybridization (aCGH).
Current Research
MIP has been used extensively in many areas of research; some of the examples of the use of this technique in recent literature are outlined below:
- Molecular inversion probe technique has been used in studying childhood brain tumors, the most common solid pediatric cancer and the leading cause of pediatric cancer mortality. Despite their high prevalence, little is known about the genetic events that contribute to the development and progression of pediatric gliomas. MIP has identified novel areas of copy number events in this cancer using minimal DNA. Identification of these events can in return lead to the understanding of the underlying mechanism of this disease.[8]
- 45 pediatric leukemia samples were analyzed for gene copy aberrations using molecular inversion probe technology. The MIP analysis identified 69 regions of recurring copy number changes, of which 41 have not been identified with other DNA microarray platforms. Copy number gains and losses were validated in 98% of clinical karyotypes and 100% of fluorescence in situ hybridization studies available.[9]
- In another study, the MIP was used to identify the association between the polymorphisms and haplotypes in the caspase-3, caspase-7, and caspase-8 genes and the risk for endometrial cancer.[10]
- A recent study has demonstrated the success of MIP for copy number variation and genotyping studies in formalin-fixed paraffin embedded samples. These banked samples, usually with extensive follow-up information, underperform or suffer high failure rates compared to fresh frozen samples because of DNA degradation and cross-linking during the fixation and processing. The study, however, successfully applied MIP to obtain high quality copy number and genotyping data from formalin-fixed paraffin embedded samples.[11]
- Molecular inversion probe technique has also been used in the field of pharmacogenomics. Genotyping of genes important in drug metabolism, excretion and transport using MIP has paved the way in understanding the patient-to-patient variability in responses to drugs.[12]
MIP Design and Optimization
Probe Design Optimization Strategies
To optimize the degree of multiplexing and the lengths of the captured regions, a number of factors should be considered when designing probes [2]:
- The sequences of the probe that are complementary to the DNA target must be specific and map only to unique regions with reasonable sequence complexity in the genome . Genomic regions containing repeats should be treated with caution.
- For all probes used in a single assay, the annealing temperatures of the two target complementary ends of the probes should be similar such that hybridization of the two ends to their targets can be achieved at the same temperature[3].
- The GC content of the genomic targets should be similar and the targets lengths variability should be restricted such that all gaps can be filled under similar elongation timeframes [2].
- The lengths of the genomic targets cannot be too long (current successful applications worked with 100 to 200bp target lengths[2]), otherwise steric effects may interfere with successful hybridization of the probes to their targets [2].
- The tags from each probe used for microarray-based captured region identification should have similar melting temperatures as well as maximal orthogonal base complexities. These ensure that all tags can be hybridized to the array under similar conditions and that cross-hybridizations are minimized, respectively.
MIP Protocol Optimization Strategies
A number of experimental conditions can be modified for optimization[2], these include:
- Hybridization and gap-fill time
- Probes, Ligase and DNA polymerase concentrations
- Enrichment of the captured target by either rolling circle amplification or linearizing the probes to perform multi-template PCR using the universal primers, common for all probes
- Captured target identification via either array-based hybridization approaches or direct sequencing of the target
These factors are critical since in one study, proper optimization strategies increased target capture efficiency from 18 to 91 percent [13].
Performance Metrics
Turner et al. 2009[2] summarized two metrics that are commonly reported in MIP-based genomic capture experiments that identify the target by sequencing.
- Capture Uniformity: analogous to recall – the fraction of genomic targets that are captured with confidence. Specifically, the relative abundance of sequence reads that are mapped to each genomic target.
- Capture Specificity: analogous to precision – the fraction of sequence reads that actually map to the genomic targets of interest.
These two metrics are directly affected by the quality of the batch of probes. To improve the results for low quality probes, higher levels of sequencing depths can be performed. The amount of sequencing scales needed nearly exponentially with decreases in uniformity and specificity.
Hardenbol et al. 2005[6] proposed a set of metrics that concern SNP genotyping using MIPs.
- Single/noise ratio: Ratio of true genotype counts over background counts
- Probe conversion rate: Number of genomic SNP loci for which probes can be designed and successfully assayed. In other words, this metric concerns the fraction of probes that produce genotyping results.
- Call rate: For a given SNP locus, the number of DNA samples whose genotypes at this locus can be measured. In other words, the number of supporting evidence for the genotype(s) assigned to the given SNP locus.
- Completeness: For the set of SNPs assayed, the total fraction of genotypes that are successfully obtained.
- Accuracy: For the set of SNPs assayed, the fraction detected genotypes that are correct. This is commonly measured by the repeatability of the results.
An inherent trade-off exists between probe conversion rate and accuracy. Removing probes that yielded incorrect genotypes increases the accuracy but decreases the probe conversion rate. In contrast, using a lenient probe acceptance threshold increases probe conversion rate but decreases the accuracy[6].
Other Genomic Partitioning Techniques
 Overview of the different Genomic Partitioning techniques
Overview of the different Genomic Partitioning techniquesTo reduce the costs from sequencing whole genomes, many methods that enrich specific genomic regions of interest have been proposed.
Technique Details Multiplex Levela Multiplex PCR[14] Target enrichment by PCR amplication of the genomic targets using multiple target-specific primer sets 102 - 103 Capture by Circularization[3][5][6][15][16] Target capture using probes containing sequences complementary to the target
Hybridization of the probes to their targets results in circularized products
Target enrichment via rolling circle amplification or PCR using universal primers104 - 105 Solution-based Capture[17] Genomic DNA shotgun fragments in solution captured by biotinylated probes with
sequences complementary to the desired targets104 - 105 Array-based Capture[18] Genomic DNA shotgun fragments captured on microarray containing spots with sequences complementary to the desired targets 105 - 106 aThe number of genomic loci that can be assayed in a single run Other Capture by Circularization Methods
Gene selector method[15]: An initial multiplex PCR step is performed to enrich the targets of interest. The PCR products are circularized upon hybridization to target-specific probes with sequences complementary to the two primers used in the PCR step.
Capture by selective circularization method[16]: The genomic DNA is digested into fragments with restriction enzymes. Using selector probes with flanking regions that are complementary to the target of interest, the digested DNA fragments are circularized upon hybridization to the selector probes.
Performance Comparisons between Genomic Partitioning Techniques
Each method demonstrates trade offs between uniformity, capture specificity, cost, scalability and availability.
- In terms of capture specificity, Capture by Circularization methods demonstrate the best results. This is due to the fact that all methods in this class require two ends of the same DNA molecule (e.g. two ends of MIP probes) to simultaneously bind to a single cognate partner molecule (e.g. genomic target region) in the proper configuration for successful ligation.
- In contrast, Capture by Circularization methods demonstrate less uniformity compared to other methods. This is because the probe design for each distinct genomic target is unique and thus the performance between individual probes may vary.
- Regarding scalability, high specificity of Capture by Circularization and Solution-based Capture methods make them the most appropriate for studies which involve large number of genomic targets and many samples. Array-based Capture techniques are appropriate for studying many genomic targets but with fewer samples due to limited resolution and specificity of microarrays. Multiplex PCR methods are most appropriate for small-scale studies due to it ease of use and availability of reagents.
- The costs associated with each technique are difficult to compare given the vast choices of designs and experimental conditions. However, for every technique, attaining a high multiplexing level where many loci are assayed simultaneously amortizes the costs.
Advantages of MIP
- Unlike some of the other genotyping techniques, the need to PCR amplify the DNA sample prior to MIP application is eliminated. This is beneficial when examining a large number of target sequences simultaneously when cross-talk between primer pairs is likely to happen[3]
- High specificity: High specificity is achieved by that
i) Unlike other highly multiplexed genotyping techniques, MIP utilizes enzymatic steps (DNA polymerization and ligation) in solution to capture specific loci, which is then followed by an amplification step. Such a combination of enzymatic steps confers a high degree of specificity on the MIP assay[19]
ii) Exonuclease treatment removes non-reacted, linear probes
iii) The tag sequences are selected in a way to increase specificity at hybridization and thus prevent cross-talk at the detection step[3]
iv) Target complementary sequences at both ends of the probe are physically limited to interact locally[3] - Built-in quality control of the signal to noise ratio: the MIP technique examines the possibility of all four bases for each SNP position. A homozygous SNP is expected to have a single signal and a heterozygous SNP to have two signals. Thus, the signal to noise ratio can be monitored using the background alleles and if a call has a suspicious signal, it can be discarded from the downstream analysis[3]
- High levels of multiplexing (on the order of 104-105 probes in one assay) can be achieved[2]
- Low amount of sample DNA (e.g. 0.2 ng/SNP [3]) is needed since the MIP probes can be applied directly to genomic DNA instead of shotgun libraries
- High concordance: trio concordance rate is found to be > 99.6%[1]
- Reproducibility: genotyping the same individual several times showed that the genotyped SNPs were concordant (99.9%)[3]
- High dynamic range: in CNV detection studies, up to 60 copies of amplified regions can be detected in the genome[19]
- Since MIP requires only 40 base-pairs of intact genomic DNA, its use in degraded samples, such as formaldehyde fixed paraffin embedded samples, may offer distinct advantages[19]
- Simple infrastructure (only common bench-top reagents and tools are required) and simple design make this technique broadly applicable in many laboratories
- The choices of the platform for identifying the captured target are very flexible such that cost-efficiency may be improved. For instance, the captured targets can be directly sequenced, bypassing the need for sequencing library construction.
Limitations of MIP
- Sensitivity and uniformity are relatively low compared to other genomic capture techniques since not all targets can be captured under the same experimental conditions for high-throughput runs that involve multiple probes. However, a recent study that used probes with longer linker regions improved uniformity [20].
- The plausible sizes of the target that can be captured are limited since
i) Large gap region leads to steric constraints[2] for the intramolecular circularization of the probe and
ii) Large gap requires longer probes be synthesized, increasing the costs. - The degree of multiplexing is constrained by the multiplexing capability of the method chosen for target identification. If array-based detection methods are used, the number of targets that can be assayed is limited by the available spots on the array.
- Since a distinct probe is needed to capture each region, it is costly to assay many regions. However, with multiplexity, the costs are amortized. For instance, at a multiplexity level of 1000, the costs become $0.01 per probe for each assay [3].
- MIP reaction conditions may require optimization, which is particularly important for assaying heterozygotic sites [13].
See also
- International HapMap Project
- Exome
- Exon trapping
- Polymerase chain reaction
- Rolling circle replication
- DNA microarray
- DNA sequencing
References
- ^ a b c d Absalan, Farnaz; Mostafa Ronaghi (2008). Molecular Inversion Probe Assay. Methods in Molecular Biology. 396. Humana Press. pp. 315–330. doi:10.1007/978-1-59745-515-2.
- ^ a b c d e f g h i j k l Turner EH, Ng SB, Nickerson DA, Shendure J (2009). "Methods for genomic partitioning". Annu Rev Genomics Hum Genet 10: 263–284. doi:10.1146/annurev-genom-082908-150112. ISBN 2908150112. PMID 19630561.
- ^ a b c d e f g h i j k l m n o p q r s Hardenbol P, Banér J, Jain M, Nilsson M, Namsaraev EA, Karlin-Neumann GA, Fakhrai-Rad H, Ronaghi M, Willis TD, Landegren U, Davis RW (2003). "Multiplexed genotyping with sequence-tagged molecular inversion probes". Nat Biotechnol 21 (6): 673–678. doi:10.1038/nbt821. PMID 12730666.
- ^ Wang Y, Moorhead M, Karlin-Neumann G, Falkowski M, Chen C, Siddiqui F, Davis RW, Willis TD, Faham M (2007). "Direct selection of human genomic loci by microarray hybridization". Nucleic Acids Res 33 (21): e183. doi:10.1093/nar/gni177. PMC 1301601. PMID 16314297. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1301601.
- ^ a b c d e f Akhras MS, Unemo M, Thiyagarajan S, Nyrén P, Davis RW, Fire AZ, Pourmand N (2007). Hall, Neil. ed. "Connector inversion probe technology: a powerful one-primer multiplex DNA amplification system for numerous scientific applications". PLoS One 2 (9): e195. doi:10.1371/journal.pone.0000915. PMC 1976392. PMID 17878950. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1976392.
- ^ a b c d e f Hardenbol P, Yu F, Belmont J, Mackenzie J, Bruckner C, Brundage T, Boudreau A, Chow S, Eberle J, Erbilgin A, Falkowski M, Fitzgerald R, Ghose S, Iartchouk O, Jain M, Karlin-Neumann G, Lu X, Miao X, Moore B, Moorhead M, Namsaraev E, Pasternak S, Prakash E, Tran K, Wang Z, Jones HB, Davis RW, Willis TD, Gibbs RA (2005). "Highly multiplexed molecular inversion probe genotyping: over 10,000 targeted SNPs genotyped in a single tube assay". Genome Res 15 (2): 269–275. doi:10.1101/gr.3185605. PMC 546528. PMID 15687290. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=546528.
- ^ a b c d e Nilsson M, Malmgren H, Samiotaki M, Kwiatkowski M, Chowdhary BP, Landegren U (1994). "Padlock probes: circularizing oligonucleotides for localized DNA detection". Science 265 (5181): 2085–2088. doi:10.1126/science.7522346. PMID 7522346.
- ^ J. D. Schiffman, Y. Wang, S. R. Vandenberg, P. G. Fisher, J. M. Ford, H. Ji and J. G. Hodgson (2008). "Molecular inversion probes (MIPs) identify novel copy number changes in pediatric gliomas". American Society of Clinical Oncology 26: 13006.
- ^ Joshua D. Schiffman, Yuker Wang, Lisa A. McPherson, Katrina Welch, Nancy Zhang, Ronald Davis, Norman J. Lacayo, Gary V. Dahl, Malek Faham, James M. Ford, and Hanlee P. Ji (2009). "Molecular inversion probes reveal patterns of 9p21 deletion and copy number aberrations in childhood leukemia". Cancer Genet Cytogenet 193 (1): 9–18. doi:10.1016/j.cancergencyto.2009.03.005. PMC 2776674. PMID 19602459. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2776674.
- ^ Xu HL, Xu WH, Cai Q, Feng M, Long J, Zheng W, Xiang YB, Shu XO (2009). "Polymorphisms and haplotypes in the caspase-3, caspase-7, and caspase-8 genes and risk for endometrial cancer: a population-based, case-control study in a Chinese population". Cancer Epidemiol Biomarkers Prev 18 (7): 2114–22. doi:10.1158/1055-9965.EPI-09-0152. PMC 2764360. PMID 19531679. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2764360.
- ^ Wang Y, Carlton VE, Karlin-Neumann G, Sapolsky R, Zhang L, Moorhead M, Wang ZC, Richardson AL, Warren R, Walther A, Bondy M, Sahin A, Krahe R, Tuna M, Thompson PA, Spellman PT, Gray JW, Mills GB, Faham M (2009). "High quality copy number and genotype data from FFPE samples using Molecular Inversion Probe (MIP) microarrays". BMC Med Genomics 2: 2–8. doi:10.1186/1755-8794-2-8. ISBN 1755879428. PMC 2649948. PMID 19228381. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2649948.
- ^ Daly TM, Dumaual CM, Miao X, Farmen MW, Njau RK, Fu DJ, Bauer NL, Close S, Watanabe N, Bruckner C, Hardenbol P, Hockett RD (2007). "Multiplex assay for comprehensive genotyping of genes involved in drug metabolism, excretion, and transport". Clin Chem 53 (7): 1222–30. doi:10.1373/clinchem.2007.086348. PMID 17510302.
- ^ a b Porreca GJ, Zhang K, Li JB, Xie B, Austin D, Vassallo SL, LeProust EM, Peck BJ, Emig CJ, Dahl F, Gao Y, Church GM, Shendure J (2007). "Multiplex amplification of large sets of human exons". Nat Methods 4 (11): 931–936. doi:10.1038/nmeth1110. PMID 17934468.
- ^ Meuzelaar LS, Lancaster O, Pasche JP, Kopal G, Brookes AJ (2007). "MegaPlex PCR: a strategy for multiplex amplification". Nat Methods 4 (10): 835–837. doi:10.1038/nmeth1091. PMID 17873887.
- ^ a b Fredriksson S, Baner J, Dahl F, Chu A, Ji H (2007). "Multiplex amplification of all coding sequences within 10 cancer genes by Gene-Collector". Nucleic Acids Res 35 (7): e47. doi:10.1093/nar/gkm078. PMC 1874629. PMID 17317684. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1874629.
- ^ a b Dahl F, Gullberg M, Stenberg J, Landegren U, Nilsson M (2005). "Multiplex amplification enabled by selective circularization of large sets of genomic DNA fragments". Nucleic Acids Res 33 (8): e71. doi:10.1093/nar/gni070. PMC 1087789. PMID 15860768. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1087789.
- ^ Bashiardes S, Veile R, Helms C, Mardis ER, Bowcock AM, Lovett M (2005). "Direct genomic selection". Nat Methods 2 (1): 63–69. doi:10.1038/nmeth0105-63. PMID 16152676.
- ^ Albert TJ, Molla MN, Muzny DM, Nazareth L, Wheeler D, Song X, Richmond TA, Middle CM, Rodesch MJ, Packard CJ, Weinstock GM, Gibbs RA (2007). "Direct selection of human genomic loci by microarray hybridization". Nat Methods 4 (11): 903–905. doi:10.1038/nmeth1111. PMID 17934467.
- ^ a b c Wang Y, Moorhead M, Karlin-Neumann G, Wang NJ, Ireland J, Lin S, Chen C, Heiser LM, Chin K, Esserman L, Gray JW, Spellman PT, Faham M (2007). "Analysis of molecular inversion probe performance for allele copy number determination". Genome Biol 8 (11): R246. doi:10.1186/gb-2007-8-11-r246. PMC 2258201. PMID 18028543. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2258201.
- ^ Krishnakumar S, Zheng J, Wilhelmy J, Faham M, Mindrinos M, Davis R (2008). "A comprehensive assay for targeted multiplex amplification of human DNA sequences". Proc Natl Acad Sci U S A 105 (27): 9296–9301. doi:10.1073/pnas.0803240105. PMC 2442818. PMID 18599465. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2442818.
External links
Categories:



Wikimedia Foundation. 2010.