- Cystic fibrosis transmembrane conductance regulator
-
Cystic fibrosis transmembrane conductance regulator (CFTR) is a protein[1] that in humans is encoded by the CFTR gene.[2]
CFTR is a ABC transporter-class ion channel that transports chloride and thiocyanate[3] ions across epithelial cell membranes. Mutations of the CFTR gene affect functioning of the chloride ion channels in these cell membranes, leading to cystic fibrosis and congenital absence of the vas deferens.
Contents
Gene
 The location of the CFTR gene on chromosome 7
The location of the CFTR gene on chromosome 7
The gene that encodes the CFTR protein is found on the human chromosome 7, on the long arm at position q31.2.[2] from base pair 116,907,253 to base pair 117,095,955. CFTR orthologs [4] have also been identified in all mammals for which complete genome data are available.
The CFTR gene has been used in animals as a nuclear DNA phylogenetic marker.[4] Large genomic sequences of this gene have been used to explore the phylogeny of the major groups of mammals,[5] and confirmed the grouping of placental orders into four major clades: Xenarthra, Afrotheria, Laurasiatheria, and Euarchonta plus Glires.
Mutations
Well over one thousand mutations have been described that can affect the CFTR gene. Such mutations can cause two genetic disorders, congenital bilateral absence of vas deferens and the more widely known disorder cystic fibrosis. Both disorders arise from the blockage of the movement of ions and, therefore, water into and out of cells. In congenital bilateral absence of vas deferens, the protein may be still functional but not at normal efficiency, this leads to the production of thick mucus, which blocks the developing vas deferens. In people with mutations giving rise to cystic fibrosis, the blockage in ion transport occurs in epithelial cells that line the passageways of the lungs, pancreas, and other organs. This leads to chronic dysfunction, disability, and a reduced life expectancy.
The most common mutation, ΔF508 results from a deletion (Δ) of three nucleotides which results in a loss of the amino acid phenylalanine (F) at the 508th position on the protein. As a result the protein does not fold normally and is more quickly degraded.
The vast majority of mutations are quite rare. The distribution and frequency of mutations varies among different populations which has implications for genetic screening and counseling.
Mutations consist of replacements, duplications, deletions or shortenings in the CFTR gene. This may result in proteins that may not function, work less effectively, are more quickly degraded, or are present in inadequate numbers.[6]
It has been hypothesized that mutations in the CFTR gene may confer a selective advantage to heterozygous individuals. Cells expressing a mutant form of the CFTR protein are resistant to invasion by the Salmonella typhi bacterium, the agent of typhoid fever, and mice carrying a single copy of mutant CFTR are resistant to diarrhea caused by cholera toxin.[citation needed]
List of common mutations
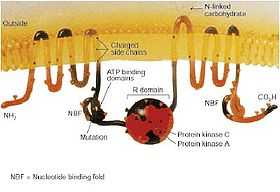
The most common mutations among caucasians are:[7]
- ΔF508
- G542X
- G551D
- N1303K
- W1282X
Structure
The CFTR gene is approximately 189 kb in length. This gene encodes the instruction to build the CFTR protein. CFTR is a glycoprotein with 1480 amino acids. The protein consists of five domains. There are two transmembrane domains, each with six spans of alpha helices. These are each connected to a nucleotide binding domain (NBD) in the cytoplasm. The first NBD is connected to the second transmembrane domain by a regulatory "R" domain that is a unique feature of CFTR, not present in other ABC transporters. The ion channel only opens when its R-domain has been phosphorylated by PKA and ATP is bound at the NBDs.[8] The carboxyl terminal of the protein is anchored to the cytoskeleton by a PDZ-interacting domain.[9]
Function
CFTR functions as a cAMP-activated ATP-gated anion channel, increasing the conductance for certain anions (e.g. Cl–) to flow down their electrochemical gradient. ATP-driven conformational changes, which in other ABC proteins fuel uphill substrate transport across cellular membranes, in CFTR open and close a gate to allow transmembrane flow of anions down their electrochemical gradient.[10] "Single CFTR channels open and close stochastically in an ATP-dependent manner, the open state catalyzing exclusively "downhill" Cl– movement at rates of millions of ions per second, orders of magnitude too high for any enzymatic pump cycle to support."[11] Essentially, CFTR is an ion channel that evolved as a 'broken' ABC transporter that leaks when in open conformation.
The CFTR is found in the epithelial cells of many organs including the lung, liver, pancreas, digestive tract, reproductive tract, and skin. Normally, the protein moves chloride and thiocyanate[12] ions (with a negative charge) out of an epithelial cell to the covering mucus. This results in an electrical gradient being formed and in the movement of (positively charged) sodium ions in the same direction as the chloride via a paracellular pathway. Due to this movement, the water potential of the mucus is reduced. This results in the movement of water out of the cell by osmosis, and therefore a more fluid mucus.
In sweat glands, CFTR defects result in reduced transport of sodium chloride and sodium thiocyanate[13] in the reabsorptive duct and saltier sweat. This was the basis of a clinically important sweat test for cystic fibrosis before genetic screening was available.[14]
Interactions
Cystic fibrosis transmembrane conductance regulator has been shown to interact with:
Related conditions
- Congenital bilateral absence of vas deferens: Males with congenital bilateral absence of the vas deferens most often have a mild mutation (a change that allows partial function of the gene) in one copy of the CFTR gene and a cystic fibrosis-causing mutation in the other copy of CFTR. As a result of these mutations, the movement of water and salt into and out of cells is disrupted. This disturbance leads to the production of a large amount of thick mucus that blocks the developing vas deferens (a tube that carries sperm from the testes) and causes it to degenerate, resulting in infertility.[28]
- Cystic fibrosis: More than 1,700 mutations in the CFTR gene have been found but the majority of these have not been associated with cystic fibrosis[citation needed]. Most of these mutations either substitute one amino acid (a building block of proteins) for another amino acid in the CFTR protein or delete a small amount of DNA in the CFTR gene. The most common mutation, called ΔF508, is a deletion (Δ) of one amino acid (phenylalanine) at position 508 in the CFTR protein. This altered protein never reaches the cell membrane because it is degraded shortly after it is made. All disease-causing mutations in the CFTR gene prevent the channel from functioning properly, leading to a blockage of the movement of salt and water into and out of cells. As a result of this blockage, cells that line the passageways of the lungs, pancreas, and other organs produce abnormally thick, sticky mucus. This mucus obstructs the airways and glands, causing the characteristic signs and symptoms of cystic fibrosis. In addition, thin mucus can be removed by cilia. However, thick mucus cannot be removed by cilia, so it traps bacteria that give rise to chronic infections.
References
- ^ Gadsby DC, Vergani P, Csanády L (March 2006). "The ABC protein turned chloride channel whose failure causes cystic fibrosis". Nature 440 (7083): 477–83. Bibcode 2006Natur.440..477G. doi:10.1038/nature04712. PMC 2720541. PMID 16554808. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2720541.
- ^ a b Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N (September 1989). "Identification of the cystic fibrosis gene: chromosome walking and jumping". Science 245 (4922): 1059–65. Bibcode 1989Sci...245.1059R. doi:10.1126/science.2772657. PMID 2772657.
- ^ Childers M, Eckel G, Himmel A, Caldwell J (2007). "A new model of cystic fibrosis pathology: lack of transport of glutathione and its thiocyanate conjugates". Med. Hypotheses 68 (1): 101–12. doi:10.1016/j.mehy.2006.06.020. PMID 16934416.
- ^ a b "OrthoMaM phylogenetic marker: CFTR coding sequence". http://www.orthomam.univ-montp2.fr/orthomam/data/cds/detailMarkers/ENSG00000001626_CFTR.xml.
- ^ Prasad A. B., Allard M. W., NISC Comparative Sequencing Program & Green E. D. (2008). "Confirming the phylogeny of mammals by use of large comparative sequence data sets". Mol. Biol. Evol. 25 (9): 1795–1808. doi:10.1093/molbev/msn104. PMC 2515873. PMID 18453548. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2515873.
- ^ Rowe SM, Miller S, Sorscher EJ (May 2005). "Cystic fibrosis". N. Engl. J. Med. 352 (19): 1992–2001. doi:10.1056/NEJMra043184. PMID 15888700.
- ^ Araújo FG, Novaes FC, Santos NP, Martins VC, Souza SM, Santos SE, Ribeiro-dos-Santos AK (January 2005). "Prevalence of deltaF508, G551D, G542X, and R553X mutations among cystic fibrosis patients in the North of Brazil". Braz. J. Med. Biol. Res. 38 (1): 11–5. doi:10.1590/S0100-879X2005000100003. PMID 15665983.
- ^ Sheppard DN, Welsh MJ (January 1999). "Structure and function of the CFTR chloride channel". Physiol. Rev. 79 (1 Suppl): S23–45. PMID 9922375.
- ^ a b Short DB, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, Stutts MJ, Milgram SL (July 1998). "An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton". J. Biol. Chem. 273 (31): 19797–801. doi:10.1074/jbc.273.31.19797. PMID 9677412.
- ^ Gadsby, D.; Vergani, P.; Csanády, L. (2006). "The ABC protein turned chloride channel whose failure causes cystic fibrosis.". Nature 440 (7083): 477–483. Bibcode 2006Natur.440..477G. doi:10.1038/nature04712. PMC 2720541. PMID 16554808. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2720541.
- ^ Miller, C. (2010). "CFTR: break a pump, make a channel". Proceedings of the National Academy of Sciences of the United States of America 107 (3): 959–960. Bibcode 2010PNAS..107..959M. doi:10.1073/pnas.0913576107. PMC 2824264. PMID 20080601. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2824264.
- ^ Moskwa P, Lorentzen D, Excoffon KJ, Zabner J, McCray PB, Nauseef WM, Dupuy C, Bánfi B (January 2007). "A novel host defense system of airways is defective in cystic fibrosis". Am. J. Respir. Crit. Care Med. 175 (2): 174–83. doi:10.1164/rccm.200607-1029OC. PMC 2720149. PMID 17082494. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2720149.
- ^ Xu Y, Szép S, Lu Z (December 2009). "The antioxidant role of thiocyanate in the pathogenesis of cystic fibrosis and other inflammation-related diseases". Proc. Natl. Acad. Sci. U.S.A. 106 (48): 20515–9. Bibcode 2009PNAS..10620515X. doi:10.1073/pnas.0911412106. PMC 2777967. PMID 19918082. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2777967.
- ^ Yonei Y, Tanaka M, Ozawa Y, Miyazaki K, Tsukada N, Inada S, Inagaki Y, Miyamoto K, Suzuki O, Okawa H (April 1992). "Primary hepatocellular carcinoma with severe hypoglycemia: involvement of insulin-like growth factors". Liver 12 (2): 90–3. PMID 1320177.
- ^ Zhang, Hui; Peters Kathryn W, Sun Fei, Marino Christopher R, Lang Jochen, Burgoyne Robert D, Frizzell Raymond A (Aug. 2002). "Cysteine string protein interacts with and modulates the maturation of the cystic fibrosis transmembrane conductance regulator". J. Biol. Chem. (United States) 277 (32): 28948–58. doi:10.1074/jbc.M111706200. ISSN 0021-9258. PMID 12039948.
- ^ Cheng, Jie; Moyer Bryan D, Milewski Michal, Loffing Johannes, Ikeda Masahiro, Mickle John E, Cutting Garry R, Li Min, Stanton Bruce A, Guggino William B (Feb. 2002). "A Golgi-associated PDZ domain protein modulates cystic fibrosis transmembrane regulator plasma membrane expression". J. Biol. Chem. (United States) 277 (5): 3520–9. doi:10.1074/jbc.M110177200. ISSN 0021-9258. PMID 11707463.
- ^ a b c Gentzsch, Martina; Cui Liying, Mengos April, Chang Xiu-Bao, Chen Jey-Hsin, Riordan John R (Feb. 2003). "The PDZ-binding chloride channel ClC-3B localizes to the Golgi and associates with cystic fibrosis transmembrane conductance regulator-interacting PDZ proteins". J. Biol. Chem. (United States) 278 (8): 6440–9. doi:10.1074/jbc.M211050200. ISSN 0021-9258. PMID 12471024.
- ^ Wang, S; Yue H, Derin R B, Guggino W B, Li M (Sep. 2000). "Accessory protein facilitated CFTR-CFTR interaction, a molecular mechanism to potentiate the chloride channel activity". Cell (UNITED STATES) 103 (1): 169–79. doi:10.1016/S0092-8674(00)00096-9. ISSN 0092-8674. PMID 11051556.
- ^ Liedtke, Carole M; Yun C H Chris, Kyle Nicole, Wang Dandan (Jun. 2002). "Protein kinase C epsilon-dependent regulation of cystic fibrosis transmembrane regulator involves binding to a receptor for activated C kinase (RACK1) and RACK1 binding to Na+/H+ exchange regulatory factor". J. Biol. Chem. (United States) 277 (25): 22925–33. doi:10.1074/jbc.M201917200. ISSN 0021-9258. PMID 11956211.
- ^ a b Park, Meeyoung; Ko Shigeru B H, Choi Joo Young, Muallem Gaia, Thomas Philip J, Pushkin Alexander, Lee Myeong-Sok, Kim Joo Young, Lee Min Goo, Muallem Shmuel, Kurtz Ira (Dec. 2002). "The cystic fibrosis transmembrane conductance regulator interacts with and regulates the activity of the HCO3- salvage transporter human Na+-HCO3- cotransport isoform 3". J. Biol. Chem. (United States) 277 (52): 50503–9. doi:10.1074/jbc.M201862200. ISSN 0021-9258. PMID 12403779.
- ^ a b Cormet-Boyaka, Estelle; Di Anke, Chang Steven Y, Naren Anjaparavanda P, Tousson Albert, Nelson Deborah J, Kirk Kevin L (Sep. 2002). "CFTR chloride channels are regulated by a SNAP-23/syntaxin 1A complex". Proc. Natl. Acad. Sci. U.S.A. (United States) 99 (19): 12477–82. Bibcode 2002PNAS...9912477C. doi:10.1073/pnas.192203899. ISSN 0027-8424. PMC 129470. PMID 12209004. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=129470.
- ^ Hegedüs, Tamás; Sessler Tamás, Scott Robert, Thelin William, Bakos Eva, Váradi András, Szabó Katalin, Homolya László, Milgram Sharon L, Sarkadi Balázs (Mar. 2003). "C-terminal phosphorylation of MRP2 modulates its interaction with PDZ proteins". Biochem. Biophys. Res. Commun. (United States) 302 (3): 454–61. doi:10.1016/S0006-291X(03)00196-7. ISSN 0006-291X. PMID 12615054.
- ^ Wang, S; Raab R W, Schatz P J, Guggino W B, Li M (May. 1998). "Peptide binding consensus of the NHE-RF-PDZ1 domain matches the C-terminal sequence of cystic fibrosis transmembrane conductance regulator (CFTR)". FEBS Lett. (NETHERLANDS) 427 (1): 103–8. doi:10.1016/S0014-5793(98)00402-5. ISSN 0014-5793. PMID 9613608.
- ^ Moyer, B D; Duhaime M, Shaw C, Denton J, Reynolds D, Karlson K H, Pfeiffer J, Wang S, Mickle J E, Milewski M, Cutting G R, Guggino W B, Li M, Stanton B A (Sep. 2000). "The PDZ-interacting domain of cystic fibrosis transmembrane conductance regulator is required for functional expression in the apical plasma membrane". J. Biol. Chem. (UNITED STATES) 275 (35): 27069–74. doi:10.1074/jbc.M004951200. ISSN 0021-9258. PMID 10852925.
- ^ Hall, R A; Ostedgaard L S, Premont R T, Blitzer J T, Rahman N, Welsh M J, Lefkowitz R J (Jul. 1998). "A C-terminal motif found in the beta2-adrenergic receptor, P2Y1 receptor and cystic fibrosis transmembrane conductance regulator determines binding to the Na+/H+ exchanger regulatory factor family of PDZ proteins". Proc. Natl. Acad. Sci. U.S.A. (UNITED STATES) 95 (15): 8496–501. Bibcode 1998PNAS...95.8496H. doi:10.1073/pnas.95.15.8496. ISSN 0027-8424. PMC 21104. PMID 9671706. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=21104.
- ^ Sun, F; Hug M J, Lewarchik C M, Yun C H, Bradbury N A, Frizzell R A (Sep. 2000). "E3KARP mediates the association of ezrin and protein kinase A with the cystic fibrosis transmembrane conductance regulator in airway cells". J. Biol. Chem. (UNITED STATES) 275 (38): 29539–46. doi:10.1074/jbc.M004961200. ISSN 0021-9258. PMID 10893422.
- ^ Naren, A P; Nelson D J, Xie W, Jovov B, Pevsner J, Bennett M K, Benos D J, Quick M W, Kirk K L (Nov. 1997). "Regulation of CFTR chloride channels by syntaxin and Munc18 isoforms". Nature (ENGLAND) 390 (6657): 302–5. Bibcode 1997Natur.390..302N. doi:10.1038/36882. ISSN 0028-0836. PMID 9384384.
- ^ Cuppens H, Cassiman JJ (2004). "CFTR mutations and polymorphisms in male infertility". Int J Androl 27 (5): 251–6. doi:10.1111/j.1365-2605.2004.00485.x. PMID 15379964.
Further reading
- Kulczycki LL, Kostuch M, Bellanti JA (2003). "A clinical perspective of cystic fibrosis and new genetic findings: relationship of CFTR mutations to genotype-phenotype manifestations". Am J Med Genet A 116 (3): 262–7. doi:10.1002/ajmg.a.10886. PMID 12503104.
- Vankeerberghen A, Cuppens H, Cassiman JJ (2002). "The cystic fibrosis transmembrane conductance regulator: an intriguing protein with pleiotropic functions". J Cyst Fibros 1 (1): 13–29. doi:10.1016/S1569-1993(01)00003-0. PMID 15463806.
- Tsui LC (1993). "Mutations and sequence variations detected in the cystic fibrosis transmembrane conductance regulator (CFTR) gene: a report from the Cystic Fibrosis Genetic Analysis Consortium". Hum. Mutat. 1 (3): 197–203. doi:10.1002/humu.1380010304. PMID 1284534.
- McIntosh I, Cutting GR (1992). "Cystic fibrosis transmembrane conductance regulator and the etiology and pathogenesis of cystic fibrosis". FASEB J. 6 (10): 2775–82. PMID 1378801.
- Drumm ML, Collins FS (1993). "Molecular biology of cystic fibrosis". Mol. Genet. Med. 3: 33–68. PMID 7693108.
- Kerem B, Kerem E (1996). "The molecular basis for disease variability in cystic fibrosis". Eur. J. Hum. Genet. 4 (2): 65–73. PMID 8744024.
- Devidas S, Guggino WB (1998). "CFTR: domains, structure, and function". J. Bioenerg. Biomembr. 29 (5): 443–51. doi:10.1023/A:1022430906284. PMID 9511929.
- Nagel G (2000). "Differential function of the two nucleotide binding domains on cystic fibrosis transmembrane conductance regulator". Biochim. Biophys. Acta 1461 (2): 263–74. doi:10.1016/S0005-2736(99)00162-5. PMID 10581360.
- Boyle MP (2003). "Unique presentations and chronic complications in adult cystic fibrosis: do they teach us anything about CFTR?". Respir. Res. 1 (3): 133–5. doi:10.1186/rr23. PMC 59552. PMID 11667976. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=59552.
- Greger R, Schreiber R, Mall M, et al. (2002). "Cystic fibrosis and CFTR". Pflugers Arch. 443 Suppl 1: S3–7. doi:10.1007/s004240100635. PMID 11845294.
- Bradbury NA (2002). "cAMP signaling cascades and CFTR: is there more to learn?". Pflugers Arch. 443 Suppl 1: S85–91. doi:10.1007/s004240100651. PMID 11845310.
- Dahan D, Evagelidis A, Hanrahan JW, et al. (2002). "Regulation of the CFTR channel by phosphorylation". Pflugers Arch. 443 Suppl 1: S92–6. doi:10.1007/s004240100652. PMID 11845311.
- Cohn JA, Noone PG, Jowell PS (2002). "Idiopathic pancreatitis related to CFTR: complex inheritance and identification of a modifier gene". J. Investig. Med. 50 (5): 247S–255S. PMID 12227654.
- Schwartz M (2003). "[Cystic fibrosis transmembrane conductance regulator (CFTR) gene: mutations and clinical phenotypes]". Ugeskr. Laeg. 165 (9): 912–6. PMID 12661515.
- Wong LJ, Alper OM, Wang BT, et al. (2004). "Two novel null mutations in a Taiwanese cystic fibrosis patient and a survey of East Asian CFTR mutations". Am. J. Med. Genet. A 120 (2): 296–8. doi:10.1002/ajmg.a.20039. PMID 12833420.
- Cuppens, Harry; Cassiman, Jean- Jacques (2005). "CFTR mutations and polymorphisms in male infertility". Int. J. Androl. 27 (5): 251–6. doi:10.1111/j.1365-2605.2004.00485.x. PMID 15379964.
- Cohn JA, Mitchell RM, Jowell PS (2005). "The impact of cystic fibrosis and PSTI/SPINK1 gene mutations on susceptibility to chronic pancreatitis". Clin. Lab. Med. 25 (1): 79–100. doi:10.1016/j.cll.2004.12.007. PMID 15749233.
- Southern KW, Peckham D (2005). "Establishing a diagnosis of cystic fibrosis". Chronic respiratory disease 1 (4): 205–10. doi:10.1191/1479972304cd044rs. PMID 16281647.
- Kandula L, Whitcomb DC, Lowe ME (2006). "Genetic issues in pediatric pancreatitis". Current gastroenterology reports 8 (3): 248–53. doi:10.1007/s11894-006-0083-8. PMID 16764792.
- Marcet B, Boeynaems JM (2007). "Relationships between cystic fibrosis transmembrane conductance regulator, extracellular nucleotides and cystic fibrosis". Pharmacol. Ther. 112 (3): 719–32. doi:10.1016/j.pharmthera.2006.05.010. PMID 16828872.
- Wilschanski M, Durie PR (2007). "Patterns of GI disease in adulthood associated with mutations in the CFTR gene". Gut 56 (8): 1153–63. doi:10.1136/gut.2004.062786. PMC 1955522. PMID 17446304. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1955522.
External links
- GeneReviews/NCBI/NIH/UW entry on CFTR-Related Disorders - Cystic Fibrosis (CF, Mucoviscidosis) and Congenital Absence of the Vas Deferens (CAVD)
- The Cystic Fibrosis Transmembrane Conductance Regulator Protein
- The Human Gene Mutation Database - CFTR Records
- Cystic Fibrosis Mutation Database
- Oak Ridge National Laboratory CFTR Information
- CFTR at OMIM (National Center for Biotechnology Information)
PDB gallery  1xmi: Crystal structure of human F508A NBD1 domain with ATP
1xmi: Crystal structure of human F508A NBD1 domain with ATP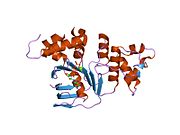 1xmj: Crystal structure of human deltaF508 human NBD1 domain with ATP
1xmj: Crystal structure of human deltaF508 human NBD1 domain with ATP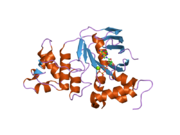 2bbo: Human NBD1 with Phe508
2bbo: Human NBD1 with Phe508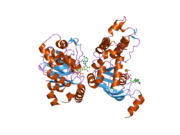 2bbs: Human deltaF508 NBD1 with three solubilizing mutations
2bbs: Human deltaF508 NBD1 with three solubilizing mutations 2bbt: Human deltaF508 NBD1 with two solublizing mutations.
2bbt: Human deltaF508 NBD1 with two solublizing mutations.Ca2+: Calcium channel Ligand-gatedNa+: Sodium channel Constitutively activeProton gatedK+: Potassium channel Kvα1-6 (1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8) · (2.1, 2.2) · (3.1, 3.2, 3.3, 3.4) · (4.1, 4.2, 4.3) · (5.1) · (6.1, 6.2, 6.3, 6.4)
Kvα7-12 (7.1, 7.2, 7.3, 7.4, 7.5) · (8.1, 8.2) · (9.1, 9.2, 9.3) · (10.1, 10.2) · (11.1/hERG, 11.2, 11.3) · (12.1, 12.2, 12.3)
Kvβ (1, 2, 3) · KCNIP (1, 2, 3, 4) · minK/ISK · minK/ISK-like · MiRP (1, 2, 3) · Shaker geneOther Cl-: Chloride channelHVCN1Generalsee also disorders
B memb: cead, trns (1A, 1C, 1F, 2A, 3A1, 3A2-3, 3D), othrA B C D E F G see also ABC transporter disorders
B memb: cead, trns (1A, 1C, 1F, 2A, 3A1, 3A2-3, 3D), othrCategories:- Human proteins
- ABC transporters





Wikimedia Foundation. 2010.