- Noonan syndrome
-
Noonan syndrome Classification and external resources 
A 12-year-old female with Noonan syndrome. Typical webbed neck. Double structural curve with rib deformity.ICD-10 Q87.1 ICD-9 759.89 OMIM 163950 DiseasesDB 29094 eMedicine article/947504 MeSH D009634 Noonan Syndrome (NS) is a relatively common autosomal dominant congenital disorder considered to be a type of dwarfism, that affects both males and females equally[1]:550. It used to be referred to as the male version of Turner's syndrome[2] (and is still sometimes described in this way);[3] however, the genetic causes of Noonan syndrome and Turner syndrome are distinct. The principal features include congenital heart defect (typically pulmonary valve stenosis), short stature, learning problems, pectus excavatum, impaired blood clotting, and a characteristic configuration of facial features including a webbed neck and a flat nose bridge. The syndrome is named after Dr. Jacqueline Noonan. It is a RASopathy.
It is believed that between approximately 1 in 1,000 and 1 in 2,500 children worldwide are born with NS. It is one of the most common genetic syndromes associated with congenital heart disease, similar in frequency to Down syndrome. However, the range and severity of features can vary greatly in patients with NS. Therefore, the syndrome is not always identified at an early age.
Contents
Characteristics
Often called a "hidden" condition, the person affected may have no obvious casual signs to the onlooker, but the problems may be many and complex. The most prevalent (common) signs are highlighted in bold with frequency listed in parentheses.
By organ system
Heart
- 2/3 of patients have one of the following heart defects
- Pulmonary Valvular Stenosis —(50%)
- Septal defects: atrial —(10%) or ventricular —(less common)
- Heart murmur
- Cardiomyopathy
Gastrointestinal system
- Failure to thrive as an infant
- Decreased appetite
- Digestive problems
- Frequent or forceful vomiting
- Swallowing difficulties
Genito-urinary system
- Cryptorchidism (undescended testicles) —
Lymphatic system
- Posterior cervical hygroma (webbed neck)
- Lymphedema (build-up of body fluid due to poor functioning of the lymphatic system)
Developmental
- Clumsiness
- Poor coordination
- Motor delay
- Mental retardation —(1/3 of patients have mild MR)
- Learning disabilities
- Speech and language delays
Musculoskeletal
- Some patients with Noonan Syndrome suffer from severe joint pain or muscle pain often with no identifiable cause
Hematologic
- Easy bruising
- Amegakaryocytic Thrombocytopenia (low platelet count)
- Blood Clotting Disorders
- Von Willebrand disease
- Prolonged activated partial thromboplastin time
- Partial deficiency of Factor VIII:C
- Partial deficiency of Factor XI:C
- Partial deficiency of Factor XII:C
- Combined Coagulation deficiencies
Neurological
Arnold-Chiari Malformation (Type 1) has been noted in some patients with Noonan Syndrome
By physical appearance
Stature and posture
- Short stature
- Cervical (neck) spine fusion
- Scoliosis
- Prominence of breast bone (pectus carinatum)
- Depression of breast bone (pectus excavatum)
- Joint contractures or tightness
- Joint hyperextensibility or looseness
- Growth retardation
- Winging of the scapula
- Hypotonia (low muscle tone)
Head
- Excess skin on the back of the neck
- Low hairline at the nape of the neck
- High hairline at the front of the head
- Large head
- Triangular face shape
- Broad forehead
- Short neck, webbed neck, posterior cervical
- Curly hair
Eyes
- Widely set eyes (hypertelorism) —(95%)
- Drooping of the eyelids (ptosis (eyelid))
- Epicanthal folds (extra fold of skin at the inner corner of the eye)
- Proptosis (bulging eyes)
- Refractive visual errors
- Inward or outward turning of the eyes (strabismus)
- Nystagmus - jerking movement of the eyes
Nose
- Small, upturned nose
Ears and hearing
- Low set ears —(over 90%)
- Backward rotated ears —(over 90%)
- Thick helix of ear (outer rim) —(over 90%)
- Incomplete folding of ears
- Chronic Otitis media (ear infections)
Mouth and speech
- Deeply grooved philtrum (top lip line) —(over 90%)
- Micrognathia (undersized lower jaw)
- High Arched palate
- Dental problems
- Articulation Difficulties
- Poor tongue control
Limbs/extremities
- Bluntly ended fingers
- Extra padding on fingers and toes
- Edema of the back of hands and tops of feet
- Cubitus valgus (elbow deformity: with abnormal turning-in)
Skin
- Lymphedema (swelling of the extremities)
- Keloids (scar hypertrophy)
- Hyperkeratosis - overdevelopment of outer skin layer
- Pigmented nevi (birthmark)
Cause
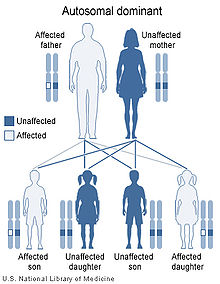 NS may be inherited in an autosomal dominant pattern with variable expression.
NS may be inherited in an autosomal dominant pattern with variable expression.
Recurrence in siblings and apparent transmission from parent to child has long suggested a genetic defect with autosomal dominant inheritance and variable expression. A person with NS has up to a 50% chance of transmitting it to a child. The fact that an affected parent is not always identified for children with NS suggests several possibilities:
- manifestations are variably expressed and could be so subtle as to go unrecognized (variable expressivity)
- a high proportion of cases represent new, sporadic mutations or
- Noonan syndrome is heterogeneous, comprising more than one similar condition of differing cause, some not inherited.
Type OMIM Gene Description NS1 163950 PTPN11 In most of the families with multiple affected members, NS maps to chromosome 12q24.1. In 2001, it was reported that approximately half of a group of patients with Noonan syndrome carried a mutation of the PTPN11 gene at that location, which encodes protein tyrosine phosphatase SHP-2.[4] The SHP2 protein is a component of several intracellular signal transduction pathways involved in embryonic development that modulate cell division, differentiation, and migration, including that mediated by the epidermal growth factor receptor. The latter pathway is important in the formation of the cardiac semilunar valves.
Chromosomal abnormalities, such as a duplication of chromosome region 12q24 encompassing gene PTPN11 can result in an apparent Noonan syndrome.[5]NS2 605275 unknown (autosomal recessive)[6] NS3 609942 KRAS Additional mutations in KRAS [7] genes have been reported to cause Noonan syndrome in a smaller percentage of individuals with the syndrome. NS4 610733 SOS1 It has recently been shown that activating mutations in SOS1 also give rise to NS.[8] Shp2 and SOS1 both have roles as positive regulators of the Ras/MAP kinase pathway suggesting that dysregulation of this pathway may play a major role in the genesis of this syndrome.[9] NS5 611553 RAF1 Additional mutations in RAF1[10] genes have been reported to cause Noonan syndrome in a smaller percentage of individuals with the syndrome. A condition known as "neurofibromatosis-Noonan syndrome" is associated with neurofibromin.[11]
Diagnosis
Despite identification of four causative genes, the diagnosis of Noonan syndrome is still based on clinical features. In other words, it is made when a physician feels that a patient has enough of the features to warrant the label indicating association. The patient can be screened for mutations in the PTPN11, SOS1, or KRAS genes, however absence of a mutation will not exclude the diagnosis as there are more as yet undiscovered genes that cause NS. The principal values of making such a diagnosis are that it guides additional medical and developmental evaluations, it excludes other possible explanations for the features, and it allows more accurate recurrence risk estimates.
History
 The oldest known case of Noonan syndrome, described in 1883 by Kobylinski.
The oldest known case of Noonan syndrome, described in 1883 by Kobylinski.Jacqueline Noonan was practicing as a pediatric cardiologist at the University of Iowa when she noticed that children with a rare type of heart defect, valvular pulmonary stenosis, often had a characteristic physical appearance with short stature, webbed neck, wide spaced eyes, and low-set ears. Both boys and girls were affected. Even though these characteristics were sometimes seen running in families, chromosomes appeared grossly normal. She studied 833 patients at the congenital heart disease clinic, looking for other congenital abnormalities, and in 1962 presented a paper: "Associated non-cardiac malformations in children with congenital heart disease". This described 9 children who in addition to congenital heart disease had characteristic faces, chest deformities and short stature. Both males and females were found to be similarly affected, and the chromosomes were apparently normal.
Dr. John Opitz, a former student of Dr. Noonan, first began to call the condition "Noonan Syndrome" when he saw children who looked like those whom Dr. Noonan had described. Dr. Noonan later produced a paper entitled "Hypertelorism with Turner Phenotype", and in 1971 at the Symposium of Cardiovascular defects, the name 'Noonan Syndrome' became officially recognized.
See also
- Turner syndrome — a different disorder which is often confused with Noonan syndrome because of several physical features that they have in common.
- Fetal alcohol syndrome — another disorder that is sometimes confused with Noonan syndrome because of some common facial features and mental retardation[12]
- Leopard syndrome— A related disorder caused by mutations in PTPN11 that are catalytically inactivating.
- Cardiofaciocutaneous syndrome — A related disorder which also affects genes encoding elements of the Ras/MAP kinase pathway.
- Dermatoglyphics
References
- ^ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- ^ Curcić-Stojković O, Nikolić L, Obradović D, Krstić A, Radić A (1978). "[Noonan's syndrome. (Male Turner's syndrome, Turner-like syndrome)]". Med Pregl 31 (7–8): 299–303. PMID 692497.
- ^ "Noonan syndrome" at Dorland's Medical Dictionary
- ^ Tartaglia M, Mehler EL, Goldberg R, et al (2001). "Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome". Nat. Genet. 29 (4): 465–8. doi:10.1038/ng772. PMID 11704759.
- ^ Shchelochkov OA et al, Am J Med Genet A, 2008 Apr 15;146A(8):1042-8
- ^ Van Der Burgt, I.; Brunner, H. (2000). "Genetic heterogeneity in Noonan syndrome: Evidence for an autosomal recessive form". American Journal of Medical Genetics 94 (1): 46. doi:10.1002/1096-8628(20000904)94:1<46::AID-AJMG10>3.0.CO;2-I. PMID 10982482.
- ^ Schubbert S, Zenker M, Rowe SL, et al (2006). "Germline KRAS mutations cause Noonan syndrome". Nat. Genet. 38 (3): 331–6. doi:10.1038/ng1748. PMID 16474405.
- ^ Roberts AE, Araki T, Swanson KD, et al (2007). "Germline gain-of-function mutations in SOS1 cause Noonan syndrome". Nat. Genet. 39 (1): 70–4. doi:10.1038/ng1926. PMID 17143285.
- ^ Bentires-Alj M, Kontaridis MI, Neel BG (2006). "Stops along the RAS pathway in human genetic disease". Nat. Med. 12 (3): 283–5. doi:10.1038/nm0306-283. PMID 16520774.
- ^ Razzaque MA, Nishizawa T, Komoike Y, et al (2007). "Germline gain-of-function mutations in RAF1 cause Noonan syndrome". Nat. Genet. 39 (8): 1013–7. doi:10.1038/ng2078. PMID 17603482.
- ^ De Luca, A.; Bottillo, I.; Sarkozy, A.; Carta, C.; Neri, C.; Bellacchio, E.; Schirinzi, A.; Conti, E. et al. (2005). "NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome". American journal of human genetics 77 (6): 1092–1101. doi:10.1086/498454. PMC 1285166. PMID 16380919. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1285166.
- ^ CDC. (2004). Fetal Alcohol Syndrome: Guidelines for Referral and Diagnosis. Can be downloaded at http://www.cdc.gov/fas/faspub.htm.
External links
- The Noonan Syndrome Support Group International - support, information & raising awareness
- Noonan syndrome at the Open Directory Project
- GeneReviews: Noonan syndrome
- Newlife Foundation: Noonan syndrome Support since 1991
Congenital abnormality · multiple abnormalities (Q87, 759.7) Craniofacial Short stature 1q21.1 deletion syndrome · Aarskog–Scott syndrome · Cockayne syndrome · Cornelia de Lange Syndrome · Dubowitz syndrome · Noonan syndrome · Robinow syndrome · Silver–Russell syndrome · Seckel syndrome · Smith-Lemli-Opitz syndrome-Turner syndromeLimbs Overgrowth Laurence-Moon-Bardet-Biedl Bardet–Biedl syndrome · Laurence-Moon syndromeCombined/other,
known locus3 (Zimmerman-Laband syndrome) · 4/13 (Fraser syndrome) · 8 (Branchio-oto-renal syndrome) · 12 (Keutel syndrome, Timothy syndrome) · 15 (Marfan syndrome) · 19 (Donohue syndrome)Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating proteinMarinesco–Sjögren syndrome · Aarskog–Scott syndrome · Juvenile primary lateral sclerosis · X-Linked mental retardation 1G protein cAMP/GNAS1: Pseudopseudohypoparathyroidism · Progressive osseous heteroplasia · Pseudohypoparathyroidism · Albright's hereditary osteodystrophy · McCune–Albright syndrome
CGL 2RAS: HRAS (Costello syndrome) · KRAS (Noonan syndrome 3, KRAS Cardiofaciocutaneous syndrome)
RAB: RAB7 (Charcot–Marie–Tooth disease) · RAB23 (Carpenter syndrome) · RAB27 (Griscelli syndrome type 2)
RHO: RAC2 (Neutrophil immunodeficiency syndrome)
ARF: SAR1B (Chylomicron retention disease) ARL13B (Joubert syndrome 8) · ARL6 (Bardet–Biedl syndrome 3)MAP kinase Other kinase/phosphatase RPS6KA3 (Coffin-Lowry syndrome) · CHEK2 (Li-Fraumeni syndrome 2) · IKBKG (Incontinentia pigmenti) · STK11 (Peutz–Jeghers syndrome) · DMPK (Myotonic dystrophy 1) · ATR (Seckel syndrome 1) · GRK1 (Oguchi disease 2) · WNK4/WNK1 (Pseudohypoaldosteronism 2)PTEN (Bannayan–Riley–Ruvalcaba syndrome, Lhermitte–Duclos disease, Cowden syndrome, Proteus-like syndrome) · MTM1 (X-linked myotubular myopathy) · PTPN11 (Noonan syndrome 1, LEOPARD syndrome, Metachondromatosis)Signal transducing adaptor proteins Other NF2 (Neurofibromatosis type II) · NOTCH3 (CADASIL) · PRKAR1A (Carney complex) · PRKAG2 (Wolff–Parkinson–White syndrome) · PRKCSH (PRKCSH Polycystic liver disease) · XIAP (XIAP2)see also intracellular signaling peptides and proteins
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkCategories:- Autosomal dominant disorders
- Syndromes
- Genodermatoses
- Enzyme defects
- RASopathies



Wikimedia Foundation. 2010.