- DNA repair
-
For the journal, see DNA Repair (journal).
 DNA damage resulting in multiple broken chromosomes
DNA damage resulting in multiple broken chromosomesDNA repair refers to a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encode its genome. In human cells, both normal metabolic activities and environmental factors such as UV light and radiation can cause DNA damage, resulting in as many as 1 million individual molecular lesions per cell per day.[1] Many of these lesions cause structural damage to the DNA molecule and can alter or eliminate the cell's ability to transcribe the gene that the affected DNA encodes. Other lesions induce potentially harmful mutations in the cell's genome, which affect the survival of its daughter cells after it undergoes mitosis. As a consequence, the DNA repair process is constantly active as it responds to damage in the DNA structure. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur, including double-strand breaks and DNA crosslinkages.[2][3]
The rate of DNA repair is dependent on many factors, including the cell type, the age of the cell, and the extracellular environment. A cell that has accumulated a large amount of DNA damage, or one that no longer effectively repairs damage incurred to its DNA, can enter one of three possible states:
- an irreversible state of dormancy, known as senescence
- cell suicide, also known as apoptosis or programmed cell death
- unregulated cell division, which can lead to the formation of a tumor that is cancerous
The DNA repair ability of a cell is vital to the integrity of its genome and thus to its normal functioning and that of the organism. Many genes that were initially shown to influence life span have turned out to be involved in DNA damage repair and protection.[4] Failure to correct molecular lesions in cells that form gametes can introduce mutations into the genomes of the offspring and thus influence the rate of evolution.
Contents
DNA damage
DNA damage, due to environmental factors and normal metabolic processes inside the cell, occurs at a rate of 1,000 to 1,000,000 molecular lesions per cell per day.[1] While this constitutes only 0.000165% of the human genome's approximately 6 billion bases (3 billion base pairs), unrepaired lesions in critical genes (such as tumor suppressor genes) can impede a cell's ability to carry out its function and appreciably increase the likelihood of tumor formation.
The vast majority of DNA damage affects the primary structure of the double helix; that is, the bases themselves are chemically modified. These modifications can in turn disrupt the molecules' regular helical structure by introducing non-native chemical bonds or bulky adducts that do not fit in the standard double helix. Unlike proteins and RNA, DNA usually lacks tertiary structure and therefore damage or disturbance does not occur at that level. DNA is, however, supercoiled and wound around "packaging" proteins called histones (in eukaryotes), and both superstructures are vulnerable to the effects of DNA damage.
Sources of damage
DNA damage can be subdivided into two main types:
- endogenous damage such as attack by reactive oxygen species produced from normal metabolic byproducts (spontaneous mutation), especially the process of oxidative deamination
- also includes replication errors
- exogenous damage caused by external agents such as
- ultraviolet [UV 200-300 nm] radiation from the sun
- other radiation frequencies, including x-rays and gamma rays
- hydrolysis or thermal disruption
- certain plant toxins
- human-made mutagenic chemicals, especially aromatic compounds that act as DNA intercalating agents
- cancer chemotherapy and radiotherapy
- viruses [5]
The replication of damaged DNA before cell division can lead to the incorporation of wrong bases opposite damaged ones. Daughter cells that inherit these wrong bases carry mutations from which the original DNA sequence is unrecoverable (except in the rare case of a back mutation, for example, through gene conversion).
Types of damage
There are five main types of damage to DNA due to endogenous cellular processes:
- oxidation of bases [e.g. 8-oxo-7,8-dihydroguanine (8-oxoG)] and generation of DNA strand interruptions from reactive oxygen species,
- alkylation of bases (usually methylation), such as formation of 7-methylguanine, 1-methyladenine, 6-O-Methylguanine
- hydrolysis of bases, such as deamination, depurination, and depyrimidination.
- "bulky adduct formation" (i.e., benzo[a]pyrene diol epoxide-dG adduct)
- mismatch of bases, due to errors in DNA replication, in which the wrong DNA base is stitched into place in a newly forming DNA strand, or a DNA base is skipped over or mistakenly inserted.
Damage caused by exogenous agents comes in many forms. Some examples are:
- UV-B light causes crosslinking between adjacent cytosine and thymine bases creating pyrimidine dimers. This is called direct DNA damage.
- UV-A light creates mostly free radicals. The damage caused by free radicals is called indirect DNA damage.
- Ionizing radiation such as that created by radioactive decay or in cosmic rays causes breaks in DNA strands. Low-level ionizing radiation may induce irreparable DNA damage (leading to replicational and transcriptional errors needed for neoplasia or may trigger viral interactions) leading to pre-mature aging and cancer.[6][7][8]
- Thermal disruption at elevated temperature increases the rate of depurination (loss of purine bases from the DNA backbone) and single-strand breaks. For example, hydrolytic depurination is seen in the thermophilic bacteria, which grow in hot springs at 40-80 °C.[9][10] The rate of depurination (300 purine residues per genome per generation) is too high in these species to be repaired by normal repair machinery, hence a possibility of an adaptive response cannot be ruled out.
- Industrial chemicals such as vinyl chloride and hydrogen peroxide, and environmental chemicals such as polycyclic hydrocarbons found in smoke, soot and tar create a huge diversity of DNA adducts- ethenobases, oxidized bases, alkylated phosphotriesters and Crosslinking of DNA just to name a few.
UV damage, alkylation/methylation, X-ray damage and oxidative damage are examples of induced damage. Spontaneous damage can include the loss of a base, deamination, sugar ring puckering and tautomeric shift.[11]
Nuclear versus mitochondrial DNA damage
In human cells, and eukaryotic cells in general, DNA is found in two cellular locations - inside the nucleus and inside the mitochondria. Nuclear DNA (nDNA) exists as chromatin during non-replicative stages of the cell cycle and is condensed into aggregate structures known as chromosomes during cell division. In either state the DNA is highly compacted and wound up around bead-like proteins called histones. Whenever a cell needs to express the genetic information encoded in its nDNA the required chromosomal region is unravelled, genes located therein are expressed, and then the region is condensed back to its resting conformation. Mitochondrial DNA (mtDNA) is located inside mitochondria organelles, exists in multiple copies, and is also tightly associated with a number of proteins to form a complex known as the nucleoid. Inside mitochondria, reactive oxygen species (ROS), or free radicals, byproducts of the constant production of adenosine triphosphate (ATP) via oxidative phosphorylation, create a highly oxidative environment that is known to damage mtDNA. A critical enzyme in counteracting the toxicity of these species is superoxide dismutase, which is present in both the mitochondria and cytoplasm of eukaryotic cells.
Senescence and apoptosis
Senescence, an irreversible state in which the cell no longer divides, is a protective response to the shortening of the chromosome ends. The telomeres are long regions of repetitive noncoding DNA that cap chromosomes and undergo partial degradation each time a cell undergoes division (see Hayflick limit).[12] In contrast, quiescence is a reversible state of cellular dormancy that is unrelated to genome damage (see cell cycle). Senescence in cells may serve as a functional alternative to apoptosis in cases where the physical presence of a cell for spatial reasons is required by the organism,[13] which serves as a "last resort" mechanism to prevent a cell with damaged DNA from replicating inappropriately in the absence of pro-growth cellular signaling. Unregulated cell division can lead to the formation of a tumor (see cancer), which is potentially lethal to an organism. Therefore, the induction of senescence and apoptosis is considered to be part of a strategy of protection against cancer.[14]
DNA damage and mutation
It is important to distinguish between DNA damage and mutation, the two major types of error in DNA. DNA damages and mutation are fundamentally different. Damages are physical abnormalities in the DNA, such as single- and double-strand breaks, 8-hydroxydeoxyguanosine residues, and polycyclic aromatic hydrocarbon adducts. DNA damages can be recognized by enzymes, and, thus, they can be correctly repaired if redundant information, such as the undamaged sequence in the complementary DNA strand or in a homologous chromosome, is available for copying. If a cell retains DNA damage, transcription of a gene can be prevented, and, thus, translation into a protein will also be blocked. Replication may also be blocked and/or the cell may die.
In contrast to DNA damage, a mutation is a change in the base sequence of the DNA. A mutation cannot be recognized by enzymes once the base change is present in both DNA strands, and, thus, a mutation cannot be repaired. At the cellular level, mutations can cause alterations in protein function and regulation. Mutations are replicated when the cell replicates. In a population of cells, mutant cells will increase or decrease in frequency according to the effects of the mutation on the ability of the cell to survive and reproduce. Although distinctly different from each other, DNA damages and mutations are related because DNA damages often cause errors of DNA synthesis during replication or repair; these errors are a major source of mutation.
Given these properties of DNA damage and mutation, it can be seen that DNA damages are a special problem in non-dividing or slowly dividing cells, where unrepaired damages will tend to accumulate over time. On the other hand, in rapidly dividing cells, unrepaired DNA damages that do not kill the cell by blocking replication will tend to cause replication errors and thus mutation. The great majority of mutations that are not neutral in their effect are deleterious to a cell’s survival. Thus, in a population of cells comprising a tissue with replicating cells, mutant cells will tend to be lost. However, infrequent mutations that provide a survival advantage will tend to clonally expand at the expense of neighboring cells in the tissue. This advantage to the cell is disadvantageous to the whole organism, because such mutant cells can give rise to cancer. Thus, DNA damages in frequently dividing cells, because they give rise to mutations, are a prominent cause of cancer. In contrast, DNA damages in infrequently dividing cells are likely a prominent cause of aging.[15]
DNA repair mechanisms
 Single-strand and double-strand DNA damage
Single-strand and double-strand DNA damageCells cannot function if DNA damage corrupts the integrity and accessibility of essential information in the genome (but cells remain superficially functional when so-called "non-essential" genes are missing or damaged). Depending on the type of damage inflicted on the DNA's double helical structure, a variety of repair strategies have evolved to restore lost information. If possible, cells use the unmodified complementary strand of the DNA or the sister chromatid as a template to recover the original information. Without access to a template, cells use an error-prone recovery mechanism known as translesion synthesis as a last resort.
Damage to DNA alters the spatial configuration of the helix, and such alterations can be detected by the cell. Once damage is localized, specific DNA repair molecules bind at or near the site of damage, inducing other molecules to bind and form a complex that enables the actual repair to take place.
Direct reversal
Cells are known to eliminate three types of damage to their DNA by chemically reversing it. These mechanisms do not require a template, since the types of damage they counteract can occur in only one of the four bases. Such direct reversal mechanisms are specific to the type of damage incurred and do not involve breakage of the phosphodiester backbone. The formation of pyrimidine dimers upon irradiation with UV light results in an abnormal covalent bond between adjacent pyrimidine bases. The photoreactivation process directly reverses this damage by the action of the enzyme photolyase, whose activation is obligately dependent on energy absorbed from blue/UV light (300–500 nm wavelength) to promote catalysis.[16] Another type of damage, methylation of guanine bases, is directly reversed by the protein methyl guanine methyl transferase (MGMT), the bacterial equivalent of which is called ogt. This is an expensive process because each MGMT molecule can be used only once; that is, the reaction is stoichiometric rather than catalytic.[17] A generalized response to methylating agents in bacteria is known as the adaptive response and confers a level of resistance to alkylating agents upon sustained exposure by upregulation of alkylation repair enzymes.[18] The third type of DNA damage reversed by cells is certain methylation of the bases cytosine and adenine.
Single-strand damage
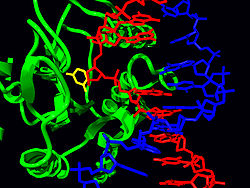 Structure of the base-excision repair enzyme uracil-DNA glycosylase. The uracil residue is shown in yellow.
Structure of the base-excision repair enzyme uracil-DNA glycosylase. The uracil residue is shown in yellow.
When only one of the two strands of a double helix has a defect, the other strand can be used as a template to guide the correction of the damaged strand. In order to repair damage to one of the two paired molecules of DNA, there exist a number of excision repair mechanisms that remove the damaged nucleotide and replace it with an undamaged nucleotide complementary to that found in the undamaged DNA strand.[17]
- Base excision repair (BER), which repairs damage to a single base caused by oxidation, alkylation, hydrolysis, or deamination. The damaged base is removed by a DNA glycosylase. The "missing tooth" is then recognized by an enzyme called AP endonuclease, which cuts the Phosphodiester bond. The missing part is then resynthesized by a DNA polymerase, and a DNA ligase performs the final nick-sealing step.
- Nucleotide excision repair (NER), which recognizes bulky, helix-distorting lesions such as pyrimidine dimers and 6,4 photoproducts. A specialized form of NER known as transcription-coupled repair deploys NER enzymes to genes that are being actively transcribed.
- Mismatch repair (MMR), which corrects errors of DNA replication and recombination that result in mispaired (but undamaged) nucleotides.
Double-strand breaks
Double-strand breaks, in which both strands in the double helix are severed, are particularly hazardous to the cell because they can lead to genome rearrangements. Three mechanisms exist to repair double-strand breaks (DSBs): non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and homologous recombination.[17] PVN Acharya noted that double-strand breaks and a "cross-linkage joining both strands at the same point is irreparable because neither strand can then serve as a template for repair. The cell will die in the next mitosis or in some rare instances, mutate."[2][3]
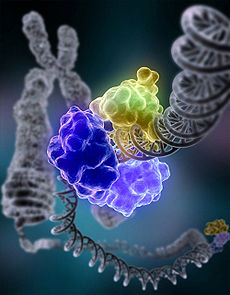 DNA ligase, shown above repairing chromosomal damage, is an enzyme that joins broken nucleotides together by catalyzing the formation of an internucleotide ester bond between the phosphate backbone and the deoxyribose nucleotides.
DNA ligase, shown above repairing chromosomal damage, is an enzyme that joins broken nucleotides together by catalyzing the formation of an internucleotide ester bond between the phosphate backbone and the deoxyribose nucleotides.In NHEJ, DNA Ligase IV, a specialized DNA ligase that forms a complex with the cofactor XRCC4, directly joins the two ends.[19] To guide accurate repair, NHEJ relies on short homologous sequences called microhomologies present on the single-stranded tails of the DNA ends to be joined. If these overhangs are compatible, repair is usually accurate.[20][21][22][23] NHEJ can also introduce mutations during repair. Loss of damaged nucleotides at the break site can lead to deletions, and joining of nonmatching termini forms translocations. NHEJ is especially important before the cell has replicated its DNA, since there is no template available for repair by homologous recombination. There are "backup" NHEJ pathways in higher eukaryotes.[24] Besides its role as a genome caretaker, NHEJ is required for joining hairpin-capped double-strand breaks induced during V(D)J recombination, the process that generates diversity in B-cell and T-cell receptors in the vertebrate immune system.[25]
Homologous recombination requires the presence of an identical or nearly identical sequence to be used as a template for repair of the break. The enzymatic machinery responsible for this repair process is nearly identical to the machinery responsible for chromosomal crossover during meiosis. This pathway allows a damaged chromosome to be repaired using a sister chromatid (available in G2 after DNA replication) or a homologous chromosome as a template. DSBs caused by the replication machinery attempting to synthesize across a single-strand break or unrepaired lesion cause collapse of the replication fork and are typically repaired by recombination.
Topoisomerases introduce both single- and double-strand breaks in the course of changing the DNA's state of supercoiling, which is especially common in regions near an open replication fork. Such breaks are not considered DNA damage because they are a natural intermediate in the topoisomerase biochemical mechanism and are immediately repaired by the enzymes that created them.
A team of French researchers bombarded Deinococcus radiodurans to study the mechanism of double-strand break DNA repair in that organism. At least two copies of the genome, with random DNA breaks, can form DNA fragments through annealing. Partially overlapping fragments are then used for synthesis of homologous regions through a moving D-loop that can continue extension until they find complementary partner strands. In the final step there is crossover by means of RecA-dependent homologous recombination.[26]
Translesion synthesis
Translesion synthesis (TLS) is a DNA damage tolerance process that allows the DNA replication machinery to replicate past DNA lesions such as thymine dimers or AP sites.[27] It involves switching out regular DNA polymerases for specialized translesion polymerases (i.e. DNA polymerase IV or V, from the Y Polymerase family), often with larger active sites that can facilitate the insertion of bases opposite damaged nucleotides. The polymerase switching is thought to be mediated by, among other factors, the post-translational modification of the replication processivity factor PCNA. Translesion synthesis polymerases often have low fidelity (high propensity to insert wrong bases) on undamaged templates relative to regular polymerases. However, many are extremely efficient at inserting correct bases opposite specific types of damage. For example, Pol η mediates error-free bypass of lesions induced by UV irradiation, whereas Pol ι introduces mutations at these sites. Pol η is known to add the first adenine across the T^T photodimer using Watson-Crick base pairing and the second adenine will be added in its syn conformation using Hoogsteen base pairing. From a cellular perspective, risking the introduction of point mutations during translesion synthesis may be preferable to resorting to more drastic mechanisms of DNA repair, which may cause gross chromosomal aberrations or cell death. In short, the process involves specialized polymerases either bypassing or repairing lesions at locations of stalled DNA replication. A bypass platform is provided to these polymerases by Proliferating cell nuclear antigen (PCNA). Under normal circumstances, PCNA bound to polymerases replicates the DNA. At a site of lesion, PCNA is ubiquitinated, or modified, by the RAD6/RAD18 proteins to provide a platform for the specialized polymerases to bypass the lesion and resume DNA replication.[28][29] After translesion synthesis, extension is required. This extension can be carried out by a replicative polymerase if the TLS is error-free, as in the case of Pol η, yet if TLS results in a mismatch, a specialized polymerase is needed to extend it; Pol ζ. Pol ζ is unique in that it can extend terminal mismatches, whereas more processive polymerases cannot. So when a lesion is encountered, the replication fork will stall, PCNA will switch from a processive polymerase to a TLS polymerase such as Pol ι to fix the lesion, then PCNA may switch to Pol ζ to extend the mismatch, and last PCNA will switch to the processive polymerase to continue replication.
Global response to DNA damage
Cells exposed to ionizing radiation, ultraviolet light or chemicals are prone to acquire multiple sites of bulky DNA lesions and double-strand breaks. Moreover, DNA damaging agents can damage other biomolecules such as proteins, carbohydrates, lipids, and RNA. The accumulation of damage, to be specific, double-strand breaks or adducts stalling the replication forks, are among known stimulation signals for a global response to DNA damage.[30] The global response to damage is an act directed toward the cells' own preservation and triggers multiple pathways of macromolecular repair, lesion bypass, tolerance, or apoptosis. The common features of global response are induction of multiple genes, cell cycle arrest, and inhibition of cell division.
DNA damage checkpoints
After DNA damage, cell cycle checkpoints are activated. Checkpoint activation pauses the cell cycle and gives the cell time to repair the damage before continuing to divide. DNA damage checkpoints occur at the G1/S and G2/M boundaries. An intra-S checkpoint also exists. Checkpoint activation is controlled by two master kinases, ATM and ATR. ATM responds to DNA double-strand breaks and disruptions in chromatin structure,[31] whereas ATR primarily responds to stalled replication forks. These kinases phosphorylate downstream targets in a signal transduction cascade, eventually leading to cell cycle arrest. A class of checkpoint mediator proteins including BRCA1, MDC1, and 53BP1 has also been identified.[32] These proteins seem to be required for transmitting the checkpoint activation signal to downstream proteins.
P53 is an important downstream target of ATM and ATR, as it is required for inducing apoptosis following DNA damage.[33] At the G1/S checkpoint, p53 functions by deactivating the CDK2/cyclin E complex. Similarly, p21 mediates the G2/M checkpoint by deactivating the CDK1/cyclin B complex[citation needed].
The prokaryotic SOS response
The SOS response is the term used to describe changes in gene expression in Escherichia coli and other bacteria in response to extensive DNA damage. The prokaryotic SOS system is regulated by two key proteins: LexA and RecA. The LexA homodimer is a transcriptional repressor that binds to operator sequences commonly referred to as SOS boxes. In Escherichia coli it is known that LexA regulates transcription of approximately 48 genes including the lexA and recA genes.[34] The SOS response is known to be widespread in the Bacteria domain, but it is mostly absent in some bacterial phyla, like the Spirochetes.[35] The most common cellular signals activating the SOS response are regions of single-stranded DNA (ssDNA), arising from stalled replication forks or double-strand breaks, which are processed by DNA helicase to separate the two DNA strands.[30] In the initiation step, RecA protein binds to ssDNA in an ATP hydrolysis driven reaction creating RecA–ssDNA filaments. RecA–ssDNA filaments activate LexA autoprotease activity, which ultimately leads to cleavage of LexA dimer and subsequent LexA degradation. The loss of LexA repressor induces transcription of the SOS genes and allows for further signal induction, inhibition of cell division and an increase in levels of proteins responsible for damage processing.
In Escherichia coli, SOS boxes are 20-nucleotide long sequences near promoters with palindromic structure and a high degree of sequence conservation. In other classes and phyla, the sequence of SOS boxes varies considerably, with different length and composition, but it is always highly conserved and one of the strongest short signals in the genome.[35] The high information content of SOS boxes permits differential binding of LexA to different promoters and allows for timing of the SOS response. The lesion repair genes are induced at the beginning of SOS response. The error-prone translesion polymerases, for example, UmuCD’2 (also called DNA polymerase V), are induced later on as a last resort.[36] Once the DNA damage is repaired or bypassed using polymerases or through recombination, the amount of single-stranded DNA in cells is decreased, lowering the amounts of RecA filaments decreases cleavage activity of LexA homodimer, which then binds to the SOS boxes near promoters and restores normal gene expression.
Eukaryotic transcriptional responses to DNA damage
Eukaryotic cells exposed to DNA damaging agents also activate important defensive pathways by inducing multiple proteins involved in DNA repair, cell cycle checkpoint control, protein trafficking and degradation. Such genome wide transcriptional response is very complex and tightly regulated, thus allowing coordinated global response to damage. Exposure of yeast Saccharomyces cerevisiae to DNA damaging agents results in overlapping but distinct transcriptional profiles. Similarities to environmental shock response indicates that a general global stress response pathway exist at the level of transcriptional activation. In contrast, different human cell types respond to damage differently indicating an absence of a common global response. The probable explanation for this difference between yeast and human cells may be in the heterogeneity of mammalian cells. In an animal different types of cells are distributed among different organs that have evolved different sensitivities to DNA damage.[36]
In general global response to DNA damage involves expression of multiple genes responsible for postreplication repair, homologous recombination, nucleotide excision repair, DNA damage checkpoint, global transcriptional activation, genes controlling mRNA decay, and many others. A large amount of damage to a cell leaves it with an important decision: undergo apoptosis and die, or survive at the cost of living with a modified genome. An increase in tolerance to damage can lead to an increased rate of survival that will allow a greater accumulation of mutations. Yeast Rev1 and human polymerase η are members of [Y family translesion DNA polymerases present during global response to DNA damage and are responsible for enhanced mutagenesis during a global response to DNA damage in eukaryotes.[30]
DNA repair and aging
Pathological effects of poor DNA repair
 DNA repair rate is an important determinant of cell pathology
DNA repair rate is an important determinant of cell pathologyExperimental animals with genetic deficiencies in DNA repair often show decreased life span and increased cancer incidence.[15] For example, mice deficient in the dominant NHEJ pathway and in telomere maintenance mechanisms get lymphoma and infections more often, and, as a consequence, have shorter lifespans than wild-type mice.[37] In similar manner, mice deficient in a key repair and transcription protein that unwinds DNA helices have premature onset of aging-related diseases and consequent shortening of lifespan.[38] However, not every DNA repair deficiency creates exactly the predicted effects; mice deficient in the NER pathway exhibited shortened life span without correspondingly higher rates of mutation.[39]
If the rate of DNA damage exceeds the capacity of the cell to repair it, the accumulation of errors can overwhelm the cell and result in early senescence, apoptosis, or cancer. Inherited diseases associated with faulty DNA repair functioning result in premature aging,[15] increased sensitivity to carcinogens, and correspondingly increased cancer risk (see below). On the other hand, organisms with enhanced DNA repair systems, such as Deinococcus radiodurans, the most radiation-resistant known organism, exhibit remarkable resistance to the double-strand break-inducing effects of radioactivity, likely due to enhanced efficiency of DNA repair and especially NHEJ.[40]
Longevity and caloric restriction
 Most life span influencing genes affect the rate of DNA damage
Most life span influencing genes affect the rate of DNA damageA number of individual genes have been identified as influencing variations in life span within a population of organisms. The effects of these genes is strongly dependent on the environment, in particular, on the organism's diet. Caloric restriction reproducibly results in extended lifespan in a variety of organisms, likely via nutrient sensing pathways and decreased metabolic rate. The molecular mechanisms by which such restriction results in lengthened lifespan are as yet unclear (see[41] for some discussion); however, the behavior of many genes known to be involved in DNA repair is altered under conditions of caloric restriction.
For example, increasing the gene dosage of the gene SIR-2, which regulates DNA packaging in the nematode worm Caenorhabditis elegans, can significantly extend lifespan.[42] The mammalian homolog of SIR-2 is known to induce downstream DNA repair factors involved in NHEJ, an activity that is especially promoted under conditions of caloric restriction.[43] Caloric restriction has been closely linked to the rate of base excision repair in the nuclear DNA of rodents,[44] although similar effects have not been observed in mitochondrial DNA.[45]
It is interesting to note that the C. elegans gene AGE-1, an upstream effector of DNA repair pathways, confers dramatically extended life span under free-feeding conditions but leads to a decrease in reproductive fitness under conditions of caloric restriction.[46] This observation supports the pleiotropy theory of the biological origins of aging, which suggests that genes conferring a large survival advantage early in life will be selected for even if they carry a corresponding disadvantage late in life.
Medicine and DNA repair modulation
Hereditary DNA repair disorders
Defects in the NER mechanism are responsible for several genetic disorders, including:
- Xeroderma pigmentosum: hypersensitivity to sunlight/UV, resulting in increased skin cancer incidence and premature aging
- Cockayne syndrome: hypersensitivity to UV and chemical agents
- Trichothiodystrophy: sensitive skin, brittle hair and nails
Mental retardation often accompanies the latter two disorders, suggesting increased vulnerability of developmental neurons.
Other DNA repair disorders include:
- Werner's syndrome: premature aging and retarded growth
- Bloom's syndrome: sunlight hypersensitivity, high incidence of malignancies (especially leukemias).
- Ataxia telangiectasia: sensitivity to ionizing radiation and some chemical agents
All of the above diseases are often called "segmental progerias" ("accelerated aging diseases") because their victims appear elderly and suffer from aging-related diseases at an abnormally young age, while not manifesting all the symptoms of old age.
Other diseases associated with reduced DNA repair function include Fanconi's anemia, hereditary breast cancer and hereditary colon cancer.
DNA repair and cancer
Inherited mutations that affect DNA repair genes are strongly associated with high cancer risks in humans. Hereditary nonpolyposis colorectal cancer (HNPCC) is strongly associated with specific mutations in the DNA mismatch repair pathway. BRCA1 and BRCA2, two famous mutations conferring a hugely increased risk of breast cancer on carriers, are both associated with a large number of DNA repair pathways, especially NHEJ and homologous recombination.
Cancer therapy procedures such as chemotherapy and radiotherapy work by overwhelming the capacity of the cell to repair DNA damage, resulting in cell death. Cells that are most rapidly dividing - most typically cancer cells - are preferentially affected. The side-effect is that other non-cancerous but rapidly dividing cells such as stem cells in the bone marrow are also affected. Modern cancer treatments attempt to localize the DNA damage to cells and tissues only associated with cancer, either by physical means (concentrating the therapeutic agent in the region of the tumor) or by biochemical means (exploiting a feature unique to cancer cells in the body).
DNA repair and evolution
The basic processes of DNA repair are highly conserved among both prokaryotes and eukaryotes and even among bacteriophage (viruses that infect bacteria); however, more complex organisms with more complex genomes have correspondingly more complex repair mechanisms.[47] The ability of a large number of protein structural motifs to catalyze relevant chemical reactions has played a significant role in the elaboration of repair mechanisms during evolution. For an extremely detailed review of hypotheses relating to the evolution of DNA repair, see.[48]
The fossil record indicates that single-cell life began to proliferate on the planet at some point during the Precambrian period, although exactly when recognizably modern life first emerged is unclear. Nucleic acids became the sole and universal means of encoding genetic information, requiring DNA repair mechanisms that in their basic form have been inherited by all extant life forms from their common ancestor. The emergence of Earth's oxygen-rich atmosphere (known as the "oxygen catastrophe") due to photosynthetic organisms, as well as the presence of potentially damaging free radicals in the cell due to oxidative phosphorylation, necessitated the evolution of DNA repair mechanisms that act specifically to counter the types of damage induced by oxidative stress.
Rate of evolutionary change
On some occasions, DNA damage is not repaired, or is repaired by an error-prone mechanism that results in a change from the original sequence. When this occurs, mutations may propagate into the genomes of the cell's progeny. Should such an event occur in a germ line cell that will eventually produce a gamete, the mutation has the potential to be passed on to the organism's offspring. The rate of evolution in a particular species (or, in a particular gene) is a function of the rate of mutation. As a consequence, the rate and accuracy of DNA repair mechanisms have an influence over the process of evolutionary change.[49]
See also
- Direct DNA damage
- Indirect DNA damage
- DNA damage theory of aging
- Accelerated aging disease
- Aging DNA
- Cell cycle
- DNA replication
- Gene therapy
- Life extension
- Human mitochondrial genetics
- Progeria
- Senescence
- The scientific journal DNA Repair under Mutation Research
References
- ^ a b Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, Darnell J. (2004). Molecular Biology of the Cell, p963. WH Freeman: New York, NY. 5th ed.
- ^ a b Acharya, PV (1971). "The isolation and partial characterization of age-correlated oligo-deoxyribo-ribonucleotides with covalently linked aspartyl-glutamyl polypeptides.". Johns Hopkins medical journal. Supplement (1): 254–60. PMID 5055816.
- ^ a b Bjorksten, J; Acharya, PV; Ashman, S; Wetlaufer, DB (1971). "Gerogenic fractions in the tritiated rat.". Journal of the American Geriatrics Society 19 (7): 561–74. PMID 5106728.
- ^ Browner, WS; Kahn, AJ; Ziv, E; Reiner, AP; Oshima, J; Cawthon, RM; Hsueh, WC; Cummings, SR. (2004). "The genetics of human longevity". Am J Med 117 (11): 851–60. doi:10.1016/j.amjmed.2004.06.033. PMID 15589490.
- ^ Roulston A, Marcellus RC, Branton PE (1999). "Viruses and apoptosis". Annu. Rev. Microbiol. 53: 577–628. doi:10.1146/annurev.micro.53.1.577. PMID 10547702. http://arjournals.annualreviews.org/doi/abs/10.1146/annurev.micro.53.1.577?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dncbi.nlm.nih.gov. Retrieved 2008-12-20.
- ^ Acharya, PVN; The Effect of Ionizing Radiation on the Formation of Age-Correlated Oligo Deoxyribo Nucleo Phospheryl Peptides in Mammalian Cells; 10th International Congress of Gerontology, Jerusalem. Abstract No. 1; January 1975. Work done while employed by Dept. of Pathology, University of Wisconsin, Madison.
- ^ Acharya, PVN; Implicatons of The Action of Low-Level Ionizing Radiation on the Inducement of Irreparable DNA Damage Leading to Mammalian Aging and Chemical Carcinogenesis.; 10th International Congress of Biochemistry, Hamburg, Germany. Abstract No. 01-1-079; July 1976. Work done while employed by Dept. of Pathology, University of Wisconsin, Madison.
- ^ Acharya, PV Narasimh; Irreparable DNA-Damage by Industrial Pollutants in Pre-mature Aging, Chemical Carcinogenesis and Cardiac Hypertrophy: Experiments and Theory; 1st International Meeting of Heads of Clinical Biochemistry Laboratories, Jerusalem, Israel. April 1977. Work conducted at Industrial Safety Institute and Behavioral Cybernetics Laboratory, University of Wisconsin, Madison.
- ^ Madigan MT, Martino JM (2006). Brock Biology of Microorganisms (11th ed.). Pearson. pp. 136. ISBN 0-13-196893-9.
- ^ Ohta, Toshihiro; Shin-ichi, Tokishita; Mochizuki, Kayo; Kawase, Jun; Sakahira, Masahide; Yamagata, Hideo (2006). "UV Sensitivity and Mutagenesis of the Extremely Thermophilic Eubacterium Thermus thermophilus HB27". Genes and Environment 28 (2): 56–61. doi:10.3123/jemsge.28.56. http://www.jstage.jst.go.jp/article/jemsge/28/2/56/_pdf.
- ^ DNA Lesions That Require Repair: http://www.ncbi.nlm.nih.gov/books/bv.fcgi?highlight=lesion&rid=mcb.table.3236
- ^ Braig, M; Schmitt, CA. (2006). "Oncogene-induced senescence: putting the brakes on tumor development". Cancer Res 66 (6): 2881–2884. doi:10.1158/0008-5472.CAN-05-4006. PMID 16540631.
- ^ Lynch, MD. (2006). "How does cellular senescence prevent cancer?". DNA Cell Biol 25 (2): 69–78. doi:10.1089/dna.2006.25.69. PMID 16460230.
- ^ Campisi J, d'Adda di Fagagna F (2007). "Cellular senescence: when bad things happen to good cells.". Rev Mol Cell Biol. 8 (9): 729–40. doi:10.1038/nrm2233. PMID 17667954.
- ^ a b c Best,BP (2009). "Nuclear DNA damage as a direct cause of aging" (PDF). Rejuvenation Research 12 (3): 199–208. doi:10.1089/rej.2009.0847. PMID 19594328. http://www.benbest.com/lifeext/Nuclear_DNA_in_Aging.pdf.
- ^ Sancar, A. (2003). "Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors". Chem Rev 103 (6): 2203–37. doi:10.1021/cr0204348. PMID 12797829.
- ^ a b c Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R. (2004). Molecular Biology of the Gene, ch. 9 and 10. Peason Benjamin Cummings; CSHL Press. 5th ed.
- ^ Volkert, MR. (1988). "Adaptive response of Escherichia coli to alkylation damage". Environ Mol Mutagen 11 (2): 241–55. doi:10.1002/em.2850110210. PMID 3278898.
- ^ Wilson, TE; Grawunder, U; Lieber, MR (1997). "Yeast DNA ligase IV mediates non-homologous DNA end joining.". Nature 388 (6641): 495–8. doi:10.1038/41365. PMID 9242411.
- ^ Moore, JK; Haber, JE (1996). "Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae". Molecular and cellular biology 16 (5): 2164–73. PMC 231204. PMID 8628283. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=231204.
- ^ Boulton, SJ; Jackson, SP (1996). "Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways". The EMBO journal 15 (18): 5093–103. PMC 452249. PMID 8890183. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=452249.
- ^ Wilson, TE; Lieber, MR (1999). "Efficient processing of DNA ends during yeast nonhomologous end joining. Evidence for a DNA polymerase beta (Pol4)-dependent pathway". The Journal of biological chemistry 274 (33): 23599–609. doi:10.1074/jbc.274.33.23599. PMID 10438542.
- ^ Budman, J; Chu, G (2005). "Processing of DNA for nonhomologous end-joining by cell-free extract". The EMBO journal 24 (4): 849–60. doi:10.1038/sj.emboj.7600563. PMC 549622. PMID 15692565. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=549622.
- ^ Wang, H; Wang, H; Perrault, AR; Takeda, Y; Qin, W; Iliakis, G. (2003). "Biochemical evidence for Ku-independent backup pathways of NHEJ". Nucleic Acids Res 31 (18): 5377–88. doi:10.1093/nar/gkg728. PMC 203313. PMID 12954774. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=203313.
- ^ Jung, D; Alt, FW (2004). "Unraveling V(D)J recombination; insights into gene regulation". Cell 116 (2): 299–311. doi:10.1016/S0092-8674(04)00039-X. PMID 14744439.
- ^ Zahradka K, Slade D, Bailone A, Sommer S, Averbeck D, Petranovic M, Lindner AB, Radman M (2006). "Reassembly of shattered chromosomes in Deinococcus radiodurans". NATURE 443 (7111): 569–573. doi:10.1038/nature05160. PMID 17006450.
- ^ Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, Walker GC (March 2009). "Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance". Microbiol. Mol. Biol. Rev. 73 (1): 134–54. doi:10.1128/MMBR.00034-08. PMC 2650891. PMID 19258535. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2650891.
- ^ http://research.chem.psu.edu/sjbgroup/projects/translesion.htm
- ^ Wang, Z (2001). "Translesion synthesis by the UmuC family of DNA polymerases". Mutat. Res. 486 (2): 59–70. doi:10.1016/S0921-8777(01)00089-1. PMID 11425512.
- ^ a b c Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. (2006). DNA Repair and Mutagenesis, part 3. ASM Press. 2nd ed.
- ^ Bakkenist, CJ; Kastan, MB. (2003). "DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation". Nature 421 (6922): 499–506. doi:10.1038/nature01368. PMID 12556884.
- ^ Wei, Qingyi; Lei Li, David Chen (2007). DNA Repair, Genetic Instability, and Cancer. World Scientific. ISBN 981-270-014-5.
- ^ Schonthal, Axel H. (2004). Checkpoint Controls and Cancer. Humana Press. ISBN 1-58829-500-1.
- ^ Janion, C. (2001). "Some aspects of the SOS response system-a critical survey". Acta Biochim Pol. 48 (3): 599–610. PMID 11833768.
- ^ a b Erill, I; Campoy, S; Barbé, J (2007). "Aeons of distress: an evolutionary perspective on the bacterial SOS response". FEMS Microbiol Rev. 31 (6): 637–656. doi:10.1111/j.1574-6976.2007.00082.x. PMID 17883408.
- ^ a b Schlacher, K; Pham, P; Cox, MM; Goodman, MF. (2006). "Roles of DNA Polymerase V and RecA Protein in SOS Damage-Induced Mutation". Chem. Rev 106 (2): 406–419. doi:10.1021/cr0404951. PMID 16464012.
- ^ Espejel, S; Martin, M; Klatt, P; Martin-Caballero, J; Flores, JM; Blasco, MA. (2004). "Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice". EMBO Rep 5 (5): 503–9. doi:10.1038/sj.embor.7400127. PMC 1299048. PMID 15105825. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1299048.
- ^ De Boer, J; Andressoo, JO; De Wit, J; Huijmans, J; Beems, RB; Van Steeg, H; Weeda, G; Van Der Horst, GT et al. (2002). "Premature aging in mice deficient in DNA repair and transcription". Science 296 (5571): 1276–9. doi:10.1126/science.1070174. PMID 11950998.
- ^ Dolle, ME; Busuttil, RA; Garcia, AM; Wijnhoven, S; van Drunen, E; Niedernhofer, LJ; der Horst, G; Hoeijmakers, JH et al. (2006). "Increased genomic instability is not a prerequisite for shortened life span in DNA repair deficient mice". Mutation Research 596 (1–2): 22–35. doi:10.1016/j.mrfmmm.2005.11.008. PMID 16472827.
- ^ Kobayashi, Y; Narumi, I; Satoh, K; Funayama, T; Kikuchi, M; Kitayama, S; Watanabe, H. (2004). "Radiation response mechanisms of the extremely radioresistant bacterium Deinococcus radiodurans". Biol Sci Space 18 (3): 134–5. PMID 15858357.
- ^ Spindler, SR. (2005). "Rapid and reversible induction of the longevity, anticancer and genomic effects of caloric restriction". Mech Ageing Dev 126 (9): 960–6. doi:10.1016/j.mad.2005.03.016. PMID 15927235.
- ^ Tissenbaum, HA; Guarente, L. (2001). "Increased dosage of a sir-2 gene extends life span in Caenorhabditis elegans". Nature 410 (6825): 227–30. doi:10.1038/35065638. PMID 11242085.
- ^ Cohen, HY; Miller, C; Bitterman, KJ; Wall, NR; Hekking, B; Kessler, B; Howitz, KT; Gorospe, M et al. (2004). "Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase". Science 305 (5682): 390–2. doi:10.1126/science.1099196. PMID 15205477.
- ^ Cabelof, DC; Yanamadala, S; Raffoul, JJ; Guo, Z; Soofi, A; Heydari, AR. (2003). "Caloric restriction promotes genomic stability by induction of base excision repair and reversal of its age-related decline". DNA Repair (Amst.) 2 (3): 295–307. doi:10.1016/S1568-7864(02)00219-7. PMID 12547392.
- ^ Stuart, JA; Karahalil, B; Hogue, BA; Souza-Pinto, NC; Bohr, VA. (2004). "Mitochondrial and nuclear DNA base excision repair are affected differently by caloric restriction". FASEB J 18 (3): 595–7. doi:10.1096/fj.03-0890fje. PMID 14734635.
- ^ Walker, DW; McColl, G; Jenkins, NL; Harris, J; Lithgow, GJ. (2000). "Evolution of lifespan in C. elegans". Nature 405 (6784): 296–7. doi:10.1038/35012693. PMID 10830948.
- ^ Cromie, GA; Connelly, JC; Leach, DR (2001). "Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans". Mol Cell. 8 (6): 1163–74. doi:10.1016/S1097-2765(01)00419-1. PMID 11779493.
- ^ O'Brien, PJ. (2006). "Catalytic promiscuity and the divergent evolution of DNA repair enzymes". Chem Rev 106 (2): 720–52. doi:10.1021/cr040481v. PMID 16464022.
- ^ Maresca, B; Schwartz, JH (2006). "Sudden origins: a general mechanism of evolution based on stress protein concentration and rapid environmental change". Anat Rec B New Anat. 289 (1): 38–46. doi:10.1002/ar.b.20089. PMID 16437551.
External links
Listen to this article (info/dl)
 This audio file was created from a revision of DNA repair dated 2005-06-17, and does not reflect subsequent edits to the article. (Audio help)More spoken articles
This audio file was created from a revision of DNA repair dated 2005-06-17, and does not reflect subsequent edits to the article. (Audio help)More spoken articles- Roswell Park Cancer Institute DNA Repair Lectures
- A comprehensive list of Human DNA Repair Genes
- 3D structures of some DNA repair enzymes
- Human DNA repair diseases
- DNA repair special interest group
- DNA Repair
- DNA Damage and DNA Repair
- Segmental Progeria
- DNA-damage repair; the good, the bad, and the ugly
Other forms of repair Other/ungrouped proteins Regulation Other/ungrouped 8-Oxoguanine • Adaptive response • Meiotic recombination checkpoint • RecF pathway
DNA helicase: BLM · WRN
FANC proteins: core protein complex (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FANCM), FANCD1, FANCD2, FANCI, FANCJ, FANCNCategories:- Aging
- Cellular processes
- Gerontology
- Molecular genetics
- Mutation
- DNA repair

Wikimedia Foundation. 2010.