- Discovery and development of cephalosporins
-
Cephalosporins are a broad class of bactericidal antibiotics that include the β-lactam ring and share a structural similarity and mechanism of action with other β-lactam antibiotics (e.g. penicillins, carbapenems and aztreonam).[1] The cephalosporins (and other β-lactams) have the ability to kill bacteria by inhibiting essential steps in the bacterial cell wall synthesis which in the end results in osmotic lysis and death of the bacterial cell.[2] Cephalosporins are widely used antibiotics because of their clinical efficiency and desirable safety profile.[3]
The cephalosporins are diverse in their antibacterial spectrum, water solubility, acid tolerability, oral bioavailability, biological half-life and other properties. Therefore the cephalosporins can be further classified into generations depending on antibacterial activity, time of invention and structural basis.
Contents
Basic structure of cephalosporins
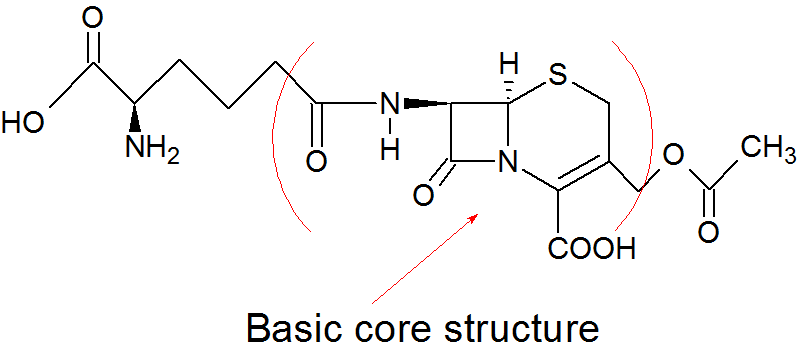
 Cephalosporin C
Cephalosporin C
The core of the basic cephalosporin molecule consists of a two ring system which includes a β-lactam ring condensed with dihydrothiazine ring. The core itself can also be referred to as 7-aminocephalosporanic acid which can be derived by hydrolysis from the natural compound cephalosporin C. Chemical compounds containing this core are relatively stable to acid hydrolysis and tolerance to β-lactamases. Cephalosporin C contains a side-chain which is derived from D-aminoadipic acid. Modification of side chains on the relevant positions has been used to create a whole new class of cephalosporin antibiotics. Modification of side-chains in position 7 of the lactam ring seems to affect the antibacterial activity while position 3 of the dihydrothiazine ring alters pharmacokinetic properties and receptor binding affinity.[4][5]
History
 A) 7-Aminocephalosporanic acid B) 7-amino-deacetoxycephalosporanic acid
A) 7-Aminocephalosporanic acid B) 7-amino-deacetoxycephalosporanic acidThe first chemical compounds of the cephalosporin group were isolated from Cephalosporium acremonium, a cephalosporin-producing fungus first discovered by Brotzu in 1948 from a sewage outfall off the Sardinian coast.[1] From crude filtrates of the Cephalosporium acremonium culture scientists got new antibacterial activity. It was noted that the crude filtrate could inhibit the growth of Staphylococcus aureus.[3] Further investigations by Abraham and Newton were made in England and isolation of culture fluids from the Sardinian fungus yielded cephalosporin P, N and C. These natural compounds were not found to be potent enough to use as antimicrobial agents but with chemical methods and removal of the natural side chain it was possible to produce 7-aminocephalosporanic acid (7-ACA) which could be further fit with unnatural side chains. 7-ACA is analogous to 6-aminopenicillanic acid(6-APA), a starting block for making several derivatives of penicillins.[1] In 1959 Abraham reported that his N-phenylacetyl derivative of cephalosporin C was much more potent against Staphylococcus aureus strains than the parent compound. This derivative was later named Cephaloram, a cephalosporin analogue of benzylpenicillin. The 7-ACA production method at Oxford, acid hydrolysis of cephalosporin C had less than 1% yield of 7-ACA which was way too low to be of commercial use. Another chemical method discovered in the laboratories of Eli Lilly had better yields of 7-ACA. That method was based on cleaving the α-aminoadipoyl side chain of cephalosporin C.[6] Further work by Robert Morin led to semisynthesis of 3-deacetoxy-7-ACA (7-ADCA) from penicillins which is convenient because penicillins can be fermented with more ease than cephalosporins. For example 7-ADCA can be semisynthesized in seven chemical reaction steps from phenoxymethylpenicillin.[1] Cephalothin, a first generation Cephalosporin for parenteral use was the first cephalosporin to become available for patients in the US. It was marketed by the pharmaceutical company Eli-lilly in 1964. Cephalothin was chosen for clinical trials from series of 7-ACA derivatives prepared at Eli-lilly. The second cephalosporin for parenteral use became available little later and was marketed in the US under the name Cephaloridine. The clinical successes of these two cephalosporins urged researchers to improve the pharmacological properties and develop more agents.[7][8] Today we are left with thousands of semisynthesized analogues of natural cephalosporin compounds based on the knowledge gained by intensive research on the chemistry of those two starting materials.[1]
Mechanism of action
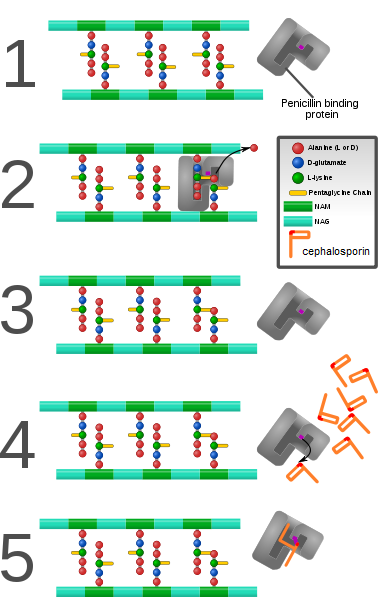
 PBP are responsible for cross-linking in the bacterial cell wall. They make peptide bonds between lysine and alanine. Cephalosporins bind into the reaction site of PBP’s rendering the enzyme unable to cross-link the bacterial wall giving bactericidal activity.
PBP are responsible for cross-linking in the bacterial cell wall. They make peptide bonds between lysine and alanine. Cephalosporins bind into the reaction site of PBP’s rendering the enzyme unable to cross-link the bacterial wall giving bactericidal activity.The bactericidal effects of β-lactam antibiotics are achieved through inhibition of the bacterial cell wall synthesis. The cell wall of both Gram-positive and Gram-negative bacteria is a tight covalently bound and cross-linked peptidoglycan network and essential for bacterial growth, cell division and cellular structure. Therefore bacteria need enzymes that can cleave the cell wall during bacterial growth and cell division. The cell wall of bacteria is built up in two steps from the outside of the cell. In the first step, molecules of disaccharide units linked with peptides on their ends are transported from the cytoplasm of the bacteria and joined together on the outside of the wall by a transglycolase. In the second step, a transpeptidase links together long polysaccharide chains which are linked together through peptide bonds. The amino acid sequence of D-alanyl-D-alanine is recognized by the transpeptidase at the end of the peptide chain. The enzyme cleaves off the alanine on the terminal end and joins the remainder to a peptide chain from an adjacent polysaccharide.[9] This transpeptidation reaction is inhibited by β-lactam antibiotics like cephalosporins. Because of this inhibition the antibiotics are most effective when the bacteria are in the logarithmic phase of growth, were then they are synthesizing the cell wall. If the bacteria are in the stationary phase of growth then there is no wall synthesizing in progress and the antibiotics have much lower effect.[3]
Although the mechanism of action for β-lactam antibiotics is not completely known they are believed to exert their mechanism of action by mimicking the structure of the transition state of the chemical reaction when the transpeptidase is bound to the D-alanyl-D-alanine sequence.[9] These proteins are often referred to as penicillin binding proteins (PBP). Opening of the β-lactam ring by a serine residue in the enzyme binding site leads to covalent binding of the antibiotic molecule with the active site of the enzyme. The result is an inactive irreversibly bound enzyme-complex which is incapable of further cell wall synthesis and the cell will die from osmotic-lysis.[2][9][10]
Drug design
Structure activity relationship
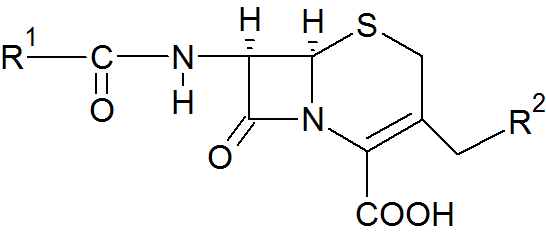
 Relevant positions of cephalosporin structure activity relationship
Relevant positions of cephalosporin structure activity relationshipThe molecular structure of cephalosporin can be altered in various ways to improve in vitro stability, anti-bacterial activity and resistance against β-lactamases. In the acidic conditions of the stomach, in vitro stability can be enhanced by the addition of an amino and a hydrogen to positions α1 and α2 of the cephalosporin structure. This results in a basic compound, an ammonium ion that is protonated in said conditions, giving us a more stable β-lactam which leads to an orally active drug. Anti-bacterial activity can be enhanced if A2 is an alkoxy group instead of a hydrogen. The 7-amino group is crucial for anti-bacterial activity. In some cases, adding a methoxy group in position A2, cephalosporin stability is enhanced toward β-lactamases. In position A1, sulfur and oxygen can be placed in the ring. Sulfur shows better anti-bacterial activity, but oxygen shows better stability towards β-lactamases. In position C6, hydrogen is crucial for biological activity. In position A3, anti-bacterial activity is greater when A3 is a 5-membered heterocycle instead of a 6-membered one. In position α1 and α2, the L-isomer is 30-40 times stabler towards β-lactamase than the D-isomer. Stability toward β-lactamase can be increased around 100-fold with the addition of methoxyoxime. Z-oxime is nearly 20,000-fold stabler than the E-oxime.[1]
Binding site
Advances in the field of recombinant protein engineering and expression, protein purification, NMR, X-ray crystallography and computational chemistry have improved the skills of drug designers to use data that have been gathered on the three dimensional structures of protein ligand complexes.[11]
Most bacterial species have various types of PBP which differ in various ways such as enzymatic function, molecular weight and the affinity for β-lactam antibiotics. There are two types of enzymes there are particularly interesting with regard to the binding site of β-lactams, PBP and β-lactamases. Target alterations in the binding site of PBP have led to high-level resistance of β-lactams among bacterias like staphylococci, enterococci and pneumococci.[12] For example, the binding site of PBP2 in Neisseria gonorrhoeae has been structurally determined and has three sequence motifs that can be seen in nearly all β-lactam interacting enzymes.
- SXXK motif located at the N-terminal end of α2 helix and includes two residues that are important for the enzyme function.
- Ser-310 : Includes a serine nucleophile that is acylated by both peptide substrate and β-lactam antibiotics.
- Lys-313 : Plays an importand role in providing the dense hydrogen bound network at the active site and is in distance of Ser 310, ASN-364 and the carbonyl backbone of Ser-362.
- SXN motif that includes Ser-362, Ser-363 and Asn-364
- KTG motif that includes Lys-497, Thr-498 and Gly-499
Research also implies that adjacent regions to the active site which differ between different PBP have significant influence on the rate of β-lactam acylation rate.[13]
Antimicrobial resistance
Bacterial resistance to the cephalosporin compounds can take place by three mechanisms.
- Modifications in target PBP
- Drug inactivation by bacterial β-lactamases
- Drug not being able to reach target PBP in the bacterial cell
Cephalosporins must get through the bacterial cell wall in order to reach the target PBP. In comparison it is easier to penetrate the cell wall of gram-positive bacteria than the cell wall of gram-negative bacteria. The cell wall structure of gram-positive bacteria is made routinely up by peptidoglycan which allows the passage of cephalosporin-sized molecules. The cell wall structure of gram-negative bacteria is more complex, composed of polysaccharides, lipids and proteins, and is harder to penetrate. Particles get through the outer membrane through water-filled channels, or porins, which are trans membrane proteins.[14] During exposure to cephalosporins the bacteria can form resistance by itself or as selection of the next generation of bacteria after reproducing itself, by mutation.[15] Bacteria species such as pneumococci and meningococci can acquire exogenous genetic material, and incorporate it into their own chromosomes which leads to antimicrobial resistance.[16] In that manner the target PBP can be altered to have their attraction for cephalosporins and other β-lactam antibiotics lowered.[17][18] The bacteria can also replace the PBP that is vulnerable to Beta-lactam antibiotics with PBP that is less vulnerable.[19] β-lactam antibiotics can be inactivated by many types of β-lactamases, which are produced by bacteria. The enzymes hydrolyze the bond between the carbon and nitrogen atom of the β-lactam ring. There are many beta lactamases which vary in substrate specificity and host range.[20][21] The enzymes active site is easily regenerated hydrolytically so it is re-usable many times, in that way can a comparatively small amount of beta-lactamases destroy a large amount of drug. Gram-positive bacteria, such as a staphylococci, have a high release of beta-lactamases into their extracellular space, where they meet the drug outside the cell wall. Gram-negative bacteria on the other hand follow a more conservative course. They secrete their beta-lactamases into the periplasmic space between the inner and outer membrane so they can't easily escape into the extracellular space, and don't have to be biosynthesized in high quantities.[1]
Drug development
This section will review the drug development from one generation to the next with emphasis on the structural differences between the generations. The generation classification system relies on dividing the cephalosporins by their chemical properties and their relative activity against gram-negative versus gram-positive bacteria.[5] [14]. From the first generation cephalosporins to the third generation there is a development from being more effective against gram-positive bacteria to being more effective against gram-negative bacteria and less effective against gram-positive bacteria respectively. However the activity returns to a balanced effectiveness against gram-negative and gram-positive bacteria in the fourth generation.[22]
Classification of cephalosporins
The cephalosporin class is very extensive so a good classification system is necessary to distinguish different cephalosporins from each other. There are few chemical and activity features that could be used for classification, for example chemical structure, side chain properties, pharmacokinetic, spectrum of activity or clinical properties. Despite these variable features the most common classification system for cephalosporins is to divide them into generations. The generation system is based on different antimicrobial activity shown by different cephalosporins.[3][4][23]
1st generation cephalosporins
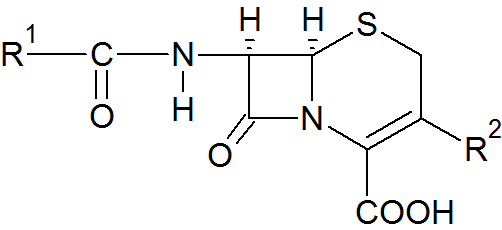
 The basic structure of first generation cephalosporins
The basic structure of first generation cephalosporinsFirst generation cephalosporins were the first cephalosporins on the market. They have good antimicrobial activity against gram-positive bacteria but limited activity against gram-negative species.[24] The chemical structures of the first generation cephalosporins are fairly simple. As an example three drugs from this class (Cephalexin, Cephradine and Cefadroxil) all have a single methyl group at position C-3. The common side groups at C-3 for first class cephalosporins are small uncharged groups like methyl.[5] The methyl group at position C-3 gives low affinity for common PBP which can in part explain the relatively low activity of these first drugs. Cefaclor however has a Cl group at position C-3 which gives it better binding to PBP and thus better antimicrobial activity. There is not an agreement on classifying Cefaclor as a first generation cephalosporin because of the Cl group at the C-3 position and therefore its improved activity, but it is often classified as such because of its C-7 side chain which is more related to the first generation then the second. All of the first generation cephalosporins have an α-amino group at position C-7. This structure makes them vulnerable to hydrolysis by β-lactamases.[5][8]
 Examples of 1st generation cephalosporins : A) Cephadrine B) Cefadroxil C) Cefalexin
Examples of 1st generation cephalosporins : A) Cephadrine B) Cefadroxil C) Cefalexin2nd generation cephalosporins
 the basic structure of 2nd generation cephalosporins
the basic structure of 2nd generation cephalosporinsEarly second generation cephalosporins are very similar in basic structure to the first generation. Loracarbef however does not have the normal dihydrothiazin ring but is a carbacephem that has a carbon atom in the ring instead of a sulfur atom making it a tetrahydropyridine ring. This chemical property gives Loracarbef better stability in plasma while retaining oral absorption characteristics and affinity for binding to PBP. The 7-phenyl-glycine makes it orally available and the chlorine at position C-3 makes it as active as Cefaclor. An important structural change in the development of second generation cephalosporins was the introduction of an α-iminomethoxy group to the C-7 side chain. This gave an increased resistance to β-lactamases due to stereochemical blocking of the beta-lactam ring. Cefuroxime was the first cephalosporin to incorporate this side chain. Another very important group in the second generation is the aminothiazole ring to the C-3 side chain. This development drastically increased binding affinity to PBP and increased antimicrobial activity. The aminothiazole ring can be seen in the structure of Cefotiam.[5][8]
 Examples of 2nd generation cephalosporins : A) Loracarbef B) Cefuroxime C) Cefotiam
Examples of 2nd generation cephalosporins : A) Loracarbef B) Cefuroxime C) Cefotiam3rd generation cephalosporins
 The basic structure of 3rd generation cephalosporins
The basic structure of 3rd generation cephalosporinsThe majority of third generation cephalosporins have the aminothiazole group at position C-7. Different groups are found at the 7-α-position like 7-α-iminohydroxy and 7-α-iminomethoxy. Ceftibuten however possesses an 7-α-ethylidene group. This group gives ceftibuten higher resistance to enhanced spectrum β-lactamases. Many of the oral third generation cephalosporins are esters of parenteral forms and are hydrolysed by esterases in the digestive tract (Cefteram-pivoxil). Some of the third generation drugs can be absorbed orally without the need of esterification. This is for example done with Cefixime and Cefdinir by putting a vinyl group in the C-3 position.[5][8]
 Examples of 3rd generation cephalosporins : A) Cefdinir B) Cefixime C) Ceftibuten
Examples of 3rd generation cephalosporins : A) Cefdinir B) Cefixime C) Ceftibuten4th generation cephalosporins
The fourth generation cephalosporins have greater activity against gram-negative bacteria than the second and third generation. This difference is attributed to them being dipolar ionic zwitterion compounds. The C-7 side chain is similar to third generation cephalosporins usually containing iminomethoxy-aminothiazole group or in the case of Cefclidine an aminothiadiazole. Because of the positively charged quaternary nitrogen in the C-3 side chain fourth generation cephalosporins can diffuse through the gram-negative bacterial membrane more readily then earlier cephalosporins. It is thought that the positive charge orients the drug molecule to the entrance of the porin channel.[25]
 Examples of 4th generation cephalosporins : A) Cefzopran B) Cefclidine C) Cefepime
Examples of 4th generation cephalosporins : A) Cefzopran B) Cefclidine C) Cefepime5th generation cephalosporins
Currently there are only two drugs in this category, Ceftobiprole and Ceftaroline. These new drugs are also the only β-lactam antibiotics that are effective against methicillin-resistant-Staphylococcus-aureus (MRSA). Ceftobiprole is a pyrrolidinone-3-ylidenemethyl cephem. The C-3 side chain was specifically designed to have a strong binding affinity to PBP2a and PBP2x. PBP2a is known to give staphylococci resistance to other β-lactam drugs and PBPx does the same for pneumococci. Ceftobiprole also has a aminothiazoylhydroxyimino side chain at the C-7 position which is known to give good resistance to β-lactamase from S. aureus. Together these active groups make Ceftobiprole bactericidal to MRSA. Ceftobiprole has poor water solubility and is therefor administered intravenously as a ester prodrug called Ceftobiprole medocaril. It is rapidly broken down into active Ceftobiprole by plasma esterases.[26] Ceftaroline was developed from the fourth generation cephalosporin Cefozopran. It retains the alkoxyimino group at position C-7 from earlier generations so it is fairly stable in the presence of many β-lactamases. Since MRSA and penicillin resistant Streptococcus pneumoniae have resistance dedicated to new types of PBP, PBP2a and PBP2x respectively, both Ceftaroline and Ceftobiprole have C-3 side chains specially engineered to bind these new PBP. In the case of Ceftaroline this side chain contains a 2-thioazolythio spacer linkage optimised for its anti-MRSA activity. Ceftaroline has low water solubility but his problem was overcome by attaching a N-phosphonoamino group to the molecule making the intravenous prodrug Ceftaroline fosamil. The prodrug is dephosphorylated in plasma to form active Ceftaroline.[27]
 5th generation cephalosporins : A) Ceftaroline phosfamil (prodrug) and B) Ceftobiprole medocaril (prodrug)
5th generation cephalosporins : A) Ceftaroline phosfamil (prodrug) and B) Ceftobiprole medocaril (prodrug)Current status
Antimicrobial resistance is the driving force for the development of new antimicrobial agents. The complexity and diversity of resistance mechanisms has defined the need for new and improved β-lactam antibiotics.[28] With their broad spectrum the cephalosporins have come to dominate β-lactam chemotherapy although they often lack oral bioavailability.[8]
On the 29th of october 2010, a new cephalosporin agent; Ceftarolin was approved by the food and drug administration (FDA). Teflaro (ceftaroline fosamil) is an injectable antibiotic prodrug to treat adults with acute bacterial skin and skin structure infections (ABSSI) and community acquired bacterial pneumonia (CABP).
Another agent; Ceftobiprole is currently being reviewed by the FDA for a new drug application, but has been approved for use in certain countries such as Canada and Switzerland.[27] In 2009 the FDA requested further clinical data from the manufacturer Basilia Pharmaceutica because of insufficient clinical testing.[29] In June 2010 the European medicines agency committee for Medicinal Products for Human Use (CHMP) recommended that the drug should not get market authorisation in Europe.
References
- ^ a b c d e f g Lemke, Thomas (2008). Foye's principles of medicinal chemistry. Philadelphia: Lippincott Williams & Wilkins. pp. 1028–1082. ISBN 978-0-7817-6879-5.
- ^ a b Klein, Lansing M. Prescott, John P. Harley, Donald A. (2005). Microbiology (6. ed. ed.). Boston, Mass.: McGraw-Hill Higher Education. ISBN 0-07-111217-0.
- ^ a b c d Singh, Jasjit; Arrieta (january 1999). "New Cephalosporins". Seminars in Pediatric Infectious Diseases 10 (1): 14–22. http://www.sciencedirect.com/science/article/pii/S1045187099800053. Retrieved 7 September 2011.
- ^ a b Goodman & Gilman's The pharmacological basis of therapeutics. (12th ed. ed.). New York: McGraw-Hill Medical. ISBN 0071624422.
- ^ a b c d e f García-Rodríguez, JA; Muñoz Bellido, JL, García Sánchez, JE (1995 Jul). "Oral cephalosporins: current perspectives.". International journal of antimicrobial agents 5 (4): 231–43. PMID 18611674.
- ^ Hamilton-Miller, J.M.T. (1 March 2008). "Development of the semi-synthetic penicillins and cephalosporins". International Journal of Antimicrobial Agents 31 (3): 189–192. doi:10.1016/j.ijantimicag.2007.11.010.
- ^ Hara, Takuji (2003). Innovation in the pharmaceutical industry : the process of drug discovery and development. Cheltenham [u.a.]: Elgar. ISBN 1843760509.
- ^ a b c d e Sader, H (1 December 1992). "Historical overview of the cephalosporin spectrum: Four generations of structural evolution". Antimicrobic Newsletter 8 (12): 75–82. doi:10.1016/0738-1751(92)90022-3.
- ^ a b c Bohlin, Gunnar Samuelsson, Lars (2009). Drugs of natural origin : a treatise of pharmacognosy (6., rev. ed. ed.). Stockholm: Apotekarsocieteten. ISBN 978-91-976510-5-9.
- ^ Miguet, Laurence; Zervosen, Astrid, Gerards, Thomas, Pasha, Farhan A., Luxen, André, Distèche-Nguyen, Martine, Thomas, Aline (8 October 2009). "Discovery of New Inhibitors of Resistant Penicillin Binding Protein (PBP) 2x by Structure-Based Virtual Screening". Journal of Medicinal Chemistry 52 (19): 5926–5936. doi:10.1021/jm900625q.
- ^ King, ed. by Frank D. (2002). Medicinal chemistry : principles and practice (2. ed. ed.). Cambridge: Royal Soc. of Chemistry. ISBN 0854046313.
- ^ Malouin, F.; Blais, J., Chamberland, S., Hoang, M., Park, C., Chan, C., Mathias, K., Hakem, S., Dupree, K., Liu, E., Nguyen, T., Dudley, M. N. (1 February 2003). "RWJ-54428 (MC-02,479), a new cephalosporin with High Affinity for Penicillin-Binding Proteins, Including PBP 2a, and Stability to Staphylococcal Beta-Lactamases". Antimicrobial Agents and Chemotherapy 47 (2): 658–664. doi:10.1128/Aac.47.2.658-664.2003.
- ^ Powell, A. J.; Tomberg, J., Deacon, A. M., Nicholas, R. A., Davies, C. (28 October 2008). "Crystal Structures of Penicillin-binding Protein 2 from Penicillin-susceptible and -resistant Strains of Neisseria gonorrhoeae Reveal an Unexpectedly Subtle Mechanism for Antibiotic Resistance". Journal of Biological Chemistry 284 (2): 1202–1212. doi:10.1074/jbc.M805761200.
- ^ Gutmann, L; Williamson, R, Collatz, E (1984 Oct). "The possible role of porins in bacterial antibiotic resistance.". Annals of internal medicine 101 (4): 554–7. PMID 6089637.
- ^ Sanders, CC; Sanders WE, Jr (1985 Mar). "Microbial resistance to newer generation β-lactam antibiotics: clinical and laboratory implications.". The Journal of infectious diseases 151 (3): 399–406. PMID 2982957.
- ^ Spratt, Brian G. (10 March 1988). "Hybrid penicillin-binding proteins in penicillin-resistant strains of Neisseria gonorrhoeae". Nature 332 (6160): 173–176. doi:10.1038/332173a0.
- ^ Fontana, R; Grossato, A, Rossi, L, Cheng, YR, Satta, G (1985 Nov). "Transition from resistance to hypersusceptibility to β-lactam antibiotics associated with loss of a low-affinity penicillin-binding protein in a Streptococcus faecium mutant highly resistant to penicillin.". Antimicrobial agents and chemotherapy 28 (5): 678–83. PMID 3853962.
- ^ Hartman, BJ; Tomasz, A (1984 May). "Low-affinity penicillin-binding protein associated with β-lactam resistance in Staphylococcus aureus.". Journal of bacteriology 158 (2): 513–6. PMID 6563036.
- ^ Handwerger, S; Tomasz, A (1986 Jan). "Alterations in penicillin-binding proteins of clinical and laboratory isolates of pathogenic Streptococcus pneumoniae with low levels of penicillin resistance.". The Journal of infectious diseases 153 (1): 83–9. PMID 3941290.
- ^ Bush, K; Jacoby, GA, Medeiros, AA (1995 Jun). "A functional classification scheme for beta-lactamases and its correlation with molecular structure.". Antimicrobial agents and chemotherapy 39 (6): 1211–33. PMID 7574506.
- ^ Livermore, DM (1995 Oct). "beta-Lactamases in laboratory and clinical resistance.". Clinical microbiology reviews 8 (4): 557–84. PMID 8665470.
- ^ Harrison, C. J.; Bratcher, D. (1 August 2008). "Cephalosporins: A Review". Pediatrics in Review 29 (8): 264–273. doi:10.1542/pir.29-8-264.
- ^ Perez-Inestrosa, E; Suau, R, Montañez, MI, Rodriguez, R, Mayorga, C, Torres, MJ, Blanca, M (2005 Aug). "Cephalosporin chemical reactivity and its immunological implications.". Current opinion in allergy and clinical immunology 5 (4): 323–30. PMID 15985814.
- ^ Kalman, D; Barriere, SL (1990). "Review of the pharmacology, pharmacokinetics, and clinical use of cephalosporins.". Texas Heart Institute journal / from the Texas Heart Institute of St. Luke's Episcopal Hospital, Texas Children's Hospital 17 (3): 203–15. PMID 15227172.
- ^ FUNGTOMC, J (1 August 1997). "Fourth-generation cephalosporins". Clinical Microbiology Newsletter 19 (17): 129–136. doi:10.1016/S0196-4399(97)82485-3.
- ^ Chahine, Elias B.; Nornoo, Adwoa O. (1 February 2011). "Ceftobiprole: The First Broad-Spectrum Anti–methicillin-resistant Staphylococcus aureus Beta-Lactam". Journal of Experimental & Clinical Medicine 3 (1): 9–16. doi:10.1016/j.jecm.2010.12.007.
- ^ a b Kaushik, Darpan; Rathi, Sudeep, Jain, Ankit (1 May 2011). "Ceftaroline: a comprehensive update". International Journal of Antimicrobial Agents 37 (5): 389–395. doi:10.1016/j.ijantimicag.2011.01.017.
- ^ Theuretzbacher, Ursula (1 October 2011). "Resistance drives antibacterial drug development". Current Opinion in Pharmacology 11 (5): 433–438. doi:10.1016/j.coph.2011.07.008.
- ^ "FDA issues ceftobiprole Complete Response Letter". Basilea Pharmaceutica Ltd.. http://www.basilea.com/News-and-Media/FDA-issues-ceftobiprole-Complete-Response-Letter/317. Retrieved 27 September 2011.
Drug design steps in design Case studies of discovery and development of drug classes triptans · cephalosporins · antiandrogens · Bcr-Abl tyrosine kinase inhibitors · nucleoside and nucleotide reverse transcriptase inhibitors · melatonin receptor agonists · proton pump inhibitors · angiotensin receptor blockers · dual serotonin and norepinephrine reuptake inhibitors · cyclooxygenase 2 inhibitors · TRPV1 antagonistsCategories:- Cephalosporin antibiotics
- History of medicine
- SXXK motif located at the N-terminal end of α2 helix and includes two residues that are important for the enzyme function.








Wikimedia Foundation. 2010.