- Development of dipeptidyl peptidase-4 inhibitors
-
Dipeptidyl peptidase-4 inhibitors (DPP-4 inhibitors) are enzyme inhibitors that inhibit the enzyme dipeptidyl peptidase-4 (DPP-4) and are a potent treatment for type 2 diabetes. Inhibition of the DPP-4 enzyme prolongs and enhances the activity of incretins that play an important role in insulin secretion and blood glucose control regulation.[1] Type 2 diabetes is a chronic metabolic disease that can be caused by pancreas β-cell dysfunction, deficiency in insulin secretion, insulin resistance and/or increased hepatic glucose production. It is one of the fastest growing health concerns in the world. Current treatments are often inefficient at sustaining glycemic control and may cause undesirable side effects, such as weight gain and episodes of hypoglycemia. Therefore, new and more effective drugs have been developed with DPP-4 inhibitors playing a significant role.[2]
Contents
History
Since its discovery in 1967, serine protease DPP-4 has been a popular subject of research.[2] Inhibitors of DPP-4 have long been sought as tools to elucidate the functional significance of the enzyme. The first inhibitors were characterized in the late 1980s and 1990s. Each inhibitor was important to establish an early structure activity relationship (SAR) for subsequent investigation. It should be noted that the inhibitors fall into two main classes, those that interact covalent with DPP-4 and those that do not.[3] DPP-4 is a dipeptidase that selectively binds substrates that contain proline at the P1-position, thus many DPP-4 inhibitors have 5-membered heterocyclic rings that mimic proline, e.g. pyrrolidine, cyanopyrrolidine, thiazolidine and cyanothiazolidine.[4] These compounds commonly form covalent bonds to the catalytic residue Ser630.[5]
In 1994, researchers from Zeria Pharmaceuticals unveiled cyanopyrrolidines with a nitrile function group that was assumed to form an imidate with the catalytic serine. Concurrently other DPP-4 inhibitors without a nitrile group were published but they contained other serine-interacting motifs, e.g. boronic acids, phosphonates or diacyl hydroxylamines. These compounds were not as potent because of the similarity of DPP-4 and prolyl oligopeptidase (PEP) and also suffered from chemical instability. Ferring Pharmaceuticals filed for patent on two cyanopyrrolidine DPP-4 inhibitors, which they published in 1995. These compounds had excellent potency and improved chemical stability.
In 1995, Edwin B. Villhauer at Novartis started to explore N-substituted glycinyl-cyanopyrrolidines based on the fact that DPP-4 identifies N-methylglycine as a N-terminal amino acid. This group of new cyanopyrrolidines became extremely popular field of research in the following years. Some trials with dual inhibitors of DPP-4 and vasopeptidase have been represented, since vasopeptidase inhibition is believed to enhance the antidiabetic effect of DPP-4 inhibition by stimulating insulin secretion. Vasopeptidase-inhibiting motif is connected to the DPP-4 inhibitor at the N-substituent.[3][6]
DPP-4 mechanism
 Fig.1: DPP-4 inhibitors inhibit DPP-4 and thus prolong the duration of GLP-1 and GIP activity, resulting in lower blood glucose level
Fig.1: DPP-4 inhibitors inhibit DPP-4 and thus prolong the duration of GLP-1 and GIP activity, resulting in lower blood glucose level
During a meal the incretins glucagon-like peptide 1 (GLP-1) and glucose-dependent gastric inhibitory polypeptide (GIP) are released from the small intestine into the vasculature. The hormones regulate insulin secretion in a glucose-dependent manner. GLP-1 has many roles in the human body; it stimulates insulin biosynthesis, inhibits glucagon secretion, slows gastric emptying, reduces appetite and stimulates regeneration of islet β-cells. GIP and GLP-1 have extremely short plasma half-lives due to a very rapid inactivation. The enzyme responsible for the metabolism is DPP-4. Inhibition of DPP-4 leads to potentiation of endogenous GIP and GLP-1 and hence improves treatment of type 2 diabetes (Figure 1).[2][7]
DPP-4 distribution and function
 Fig.2: DPP-4 cleaves two amino acids from the N-terminal end of peptides, such as GLP-1.
Fig.2: DPP-4 cleaves two amino acids from the N-terminal end of peptides, such as GLP-1.DPP-4 selectively cleaves two amino acids from peptides, such as GLP-1 and GIP, which have proline or alanine in the second position (Figure 2). At the active site of the protease there is a characteristic motif of three amino acids, Asp-His-Ser. DPP-4 is the CD26 T-cell-activating antigen, which is widely distributed in human organs and tissue. Tissues, which strongly express DPP-4, include the exocrine pancreas, sweat glands, salivary and mammary glands, thymus, lymph nodes, biliary tract, kidney, liver, placenta, uterus, prostate, skin and the capillary bed of the gut mucosa where most GLP-1 is inactivated locally. DPP-4 is attached to the plasma membrane of the endothelia of almost all organs in the body. It is also present in body fluids, such as blood plasma and cerebrospinal fluid, in a soluble form. DPP-4 inactivates GLP-1 and GIP very rapidly. Regarding GIP and GLP-1, alanine and proline are crucial for biological activity, so elimination of these amino acids leads to formation of metabolites that are inactive. Thus, preventing the degradation of the incretin hormones GIP and GLP-1 by inhibition of DPP-4 is an exciting therapeutic strategy.[1][8]
DPP-4 characteristics
Since DPP-4 is a protease, it is not unexpected that inhibitors would likely have a peptide nature and this theme has carried through to contemporary research.[3]
Structure
X-ray structures of DPP-4 that have been published since 2003 give rather detailed information about the structural characteristics of the binding site. Many structurally diverse DPP-4 inhibitors have been discovered and it is not that surprising considering the properties of the binding site:[9]
1. A deep lipophilic pocket combined with several exposed aromatic side chains for achieving high affinity small molecule binding.
2. A significant solvent access that makes it possible to tune the physico-chemical properties of the inhibitors that leads to better pharmacokinetic behavior.
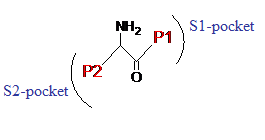
DPP-4 is a 766-amino acid transmembrane glycoprotein that belongs to the prolyloligopeptidase family. It consists of three parts; a cytoplasmic tail, a transmembrane region and an extracellular part. The extracellular part is divided into a catalytic domain and an eight-bladed β-propeller domain. The latter contributes to the inhibitor binding site. The catalytic domain shows an α/β-hydrolase fold and contains the catalytic triad Ser630 - Asp708 - His740. The S1-pocket is very hydrophobic and is composed of the side chains: Tyr631, Val656, Trp662, Tyr666 and Val711. Existing X-ray structures show that there is not much difference in size and shape of the pocket that indicates that the S1-pocket has high specificity for proline residues[8][9]
Binding site
 Fig.3: The key interactions between the ligand and DPP-4 complex. The ligand's basic amine forms a hydrogen bonding network. The nitrile reacts with the catalytic active serine and forms an imidate adduct
Fig.3: The key interactions between the ligand and DPP-4 complex. The ligand's basic amine forms a hydrogen bonding network. The nitrile reacts with the catalytic active serine and forms an imidate adductDPP-4 inhibitors usually have an electrophilic group that can interact with the hydroxyl of the catalytic serine in the active binding site (Figure 3). Frequently that group is a nitrile group but can also be boronic acid or diphenyl phosphonate. This electrophilic group can bind to the imidate complex[disambiguation needed
 ] with covalent bonds and slow, tight-binding kinetics but this group is also responsible for stability issues due to reactions with the free amino group of the P2-amino acid. Therefore inhibitors without the electrophilic group have also been developed, but these molecules have shown toxicity due to affinity to other dipeptidyl peptidases, e.g. DPP-2, DPP-8 and DPP-9.[10]
] with covalent bonds and slow, tight-binding kinetics but this group is also responsible for stability issues due to reactions with the free amino group of the P2-amino acid. Therefore inhibitors without the electrophilic group have also been developed, but these molecules have shown toxicity due to affinity to other dipeptidyl peptidases, e.g. DPP-2, DPP-8 and DPP-9.[10]DPP-4 inhibitors span diverse structural types. In 2007 few of the most potent compounds contain a proline mimetic cyanopyrrolidine P1 group. This group enhances the potency, probably due to a transient covalent trapping of the nitrile group by the active site Ser630 hydroxyl, leading to delayed dissociation and slow tight binding of certain inhibitors. When these potency enhancements were achieved, some chemical stability issues were noted and more advanced molecules had to be made. To avoid these stability issues, the possibility to exclude the nitrile group was investigated. Amino acids with aryl or polar side chains did not show appreciable DPP-4 inhibition and in fact, all compounds without the nitrile group in this research suffered a 20 to 50-fold loss of potency corresponding to the compounds containing the nitrile group.[11]
Discovery and development
It is important to find a fast and accurate system to discover new DPP-4 inhibitors with ideal therapeutic profiles. High throughput screening (HTS) usually gives low hit rates in identifying the inhibitors but virtual screening (VS) can give higher rates. VS has for example been used to screen for small primary aliphatic amines to identify fragments that could be placed in S1 and S2 sites of DPP-4. On the other hand, these fragments were not very potent and therefore identified as a starting point to design better ones. Three-dimensional models can provide a useful tool for designing novel DPP-4 inhibitors. Pharmacophore models have been made based on key chemical features of compounds with DPP-4 inhibitory activity. These models can provide a hypothetical picture of the primary chemical feature responsible for inhibitory activity.[5] The first DPP-4 inhibitors were reversible inhibitors and came with bad side effects because of low selectivity. Researchers suspected that inhibitors with short half-lives would be preferred in order to minimize possible side effects. However, since clinical trials showed the opposite, the latest DPP-4 inhibitors have a long-lasting effect. One of the first reported DPP-4 inhibitor was P32/98 from Merck. It used thiazolidide as the P1-substitute and was the first DPP-4 inhibitor that showed effects in both animals and humans but it was not developed to a market drug due to side effects. Another old inhibitor is DPP-728 from Novartis, where 2-cyanopyrrolidine is used as the P1-substitute. The addition of the cyano group generally increases the potency. Therefore, researchers' attention was directed to those compounds. Usually, DPP-4 inhibitors are either substrate-like or non-substrate-like.[12]
 Fig.4: A generic structure of a substrate-like inhibitor
Fig.4: A generic structure of a substrate-like inhibitorSubstrate-like inhibitors
Substrate-like inhibitors (Figure 4) are more common than the non-substrate-likes. They bind either covalently or non-covalently and have a basic structure where the P1-substituent occupies the S1-pocket and the P2-substituent occupies the S2-pocket. Usually they contain a proline mimetic that occupies the S1-pocket. Large substituents on the 2-cyanopyrrolidine ring are normally not tolerated since the S1-pocket is quite small.[12] Since DPP-4 is identical with the T-cell activation marker CD26 and DPP-4 inhibitors are known to inhibit T-cell proliferation, these compounds were initially thought to be potential immunomodulators. When the function against type 2 diabetes was discovered, the cyanopyrrolidines became a highly popular research material. A little later vildagliptin and saxagliptin, which are the most developed cyanopyrrolidine DPP-4 inhibitors to date, were discovered.[6]
Cyanopyrrolidines
Cyanopyrrolidines have two key interactions to the DPP-4 complex:[6]
1. Nitrile in the position of the scissile bond of the peptidic substrate that is important for high potency. The nitrile group forms reversible covalent bonds with the catalytically active serine hydroxyl (Ser630), i.e. cyanopyrrolidines are competitive inhibitors with slow dissociation kinetics.
2. Hydrogen bonding network between the protonated amino group and a negatively charged region of the protein surface, Glu205, Glu206 and Tyr662. All cyanopyrrolidines have basic, primary or secondary amine, which makes this network possible but these compounds usually drop in potency if these amines are changed. Nonetheless, two patent applications unveil that the amino group can be changed, i.e. replaced by a hydrazine, but it is claimed that these compounds do not only act via DPP-4 inhibition but also prevent diabetic vascular complications by acting as a radical scavenger.
Structure-activity relationship (SAR)
Important structure-activity relationship:[11]
1. Strict steric constraint exists around the pyrrolidine ring of cyanopyrrolidine-based inhibitors, with only hydrogen, fluoro, acetylene, nitrile, or methano substitution permitted.
2. Presence of a nitrile moiety on the pyrrolidine ring is critical to achieving potent activity
Also, systematic SAR investigation has shown that the ring size and stereochemistry for the P2 position is quite conditioned. A 5-membered ring and L-configuration has shown better results than a 4-membered or 6-membered ring with D-configuration. Only minor changes on the pyrrolidine ring can be tolerated since the good fit of the ring with the hydrophobic S1 pocket is very important for high affinity. Some trials have been made, e.g. by replacing the pyrrolidine with a thiazoline. That led to improved potency but also loss of chemical stability. Efforts to improve chemical stability often led to loss of specificity because of interactions with DPP-8 and DPP-9. These interactions have been connected with increased toxicity and mortality in animals. There are strict limitations in the P1 position and hardly any changes are tolerated, on the other hand a variety of changes can be made in the P2 position. In fact, substitution with quite big branched side chains, e.g. tert-butylglycin, normally increased activity and chemical stability, which could lead to longer-lasting inhibition of the DPP-4 enzyme. It has also been noted that biaryl-based side-chains can also give highly active inhibitors. It was originally believed that only lipophilic substitution would be tolerated. Now it is stated that also the substitution of polar negatively charged side-chains as well as hydrophilic substitution can lead to excellent inhibitory activity.[6]
Chemical stability
 Fig.5: Trans-rotamers are more stable then cis-rotamers. Cis-rotamers undergo intramolecular cyclisation.
Fig.5: Trans-rotamers are more stable then cis-rotamers. Cis-rotamers undergo intramolecular cyclisation.In general, DPP-4 inhibitors are not very stable compounds. Therefore, many researchers focus on enhancing the stability for cyanopyrrolidines. The most widespread technique to improve chemical stability is to incorporate a steric bulk. The two cyanopyrrolidines that have been most pronounced, vildagliptin and saxagliptin, were created in this manner. K579 is a DPP-4 inhibitor discovered by researchers at Kyowa Hakko Kyogo. It had improved not only chemical stability but also a longer-lasting action. That long-lasting action was most likely due to slow dissociation of the enzyme-inhibitor complex and an active oxide metabolite that undergoes enterohepatic circulation. The discovery of the active oxide was in fact a big breakthrough as it led to the development of vildagliptin and saxagliptin. One major problem in DPP-4 inhibitor stability is intramolecular cyclisation. The precondition for the intramolecular cyclisation is the conversion of the trans-rotamer, which is the DPP-4 binding rotamer(Figure 5). Thus, preventing this conversion will increase stability. This prevention was successful when incorporating an amide group into a ring, creating a compound that kept the DPP-4 inhibitory activity that, did not undergo the intramolecular cyclisation and was even more selective over different DPP enzymes. It has also been reported that a cyanoazetidine in the P1 position and a β-amino acid in the P2 position increased stability.[6]
Vildagliptin
Vildagliptin (Galvus)(Figure 6) was first synthesized in May 1998 and was named after Edwin B. Villhauer. It was discovered when researchers at Novartis examined adamantyl derivatives that had proven to be very potent. The adamantyl group worked as a steric bulk and slowed intramolecular cyclization while increasing chemical stability. Furthermore, the primary metabolites were highly active. To avoid additional chiral center a hydroxylation at the adamantyl ring was carried out (Figure 5). The product, vildagliptin, was even more stable, undergoing intramolecular cyclisation 30-times slower, and having high DPP-4 inhibitory activity and longer-lasting pharmacodynamic effect.[6]
Saxagliptin
 Fig.6: The basic structure of cyanopyrrolidines compared with vildagliptin, saxagliptin, and denagliptin
Fig.6: The basic structure of cyanopyrrolidines compared with vildagliptin, saxagliptin, and denagliptinResearchers at Bristol-Myers Squibb found that increased steric bulk of the N-terminal amino acid side-chain led to increased stability. To additionally increase stability the trans-rotamer was stabilized with a cis-4,5-methano substitution of the pyrrolidine ring, resulting in an intramolecular van-der-Waals interaction, thus preventing intramolecular cyclisation. Because of that increased stability, the researchers continued their investigation on cis-4,5-methano cyanopyrrolidines and came across with a new adamantyl derivative, which showed extraordinary ex vivo DPP-4 inhibition in rat plasma. Also noted, high microsomal turnover rate which indicated that the derivative was quickly converted to an active metabolite. After hydroxylation on the adamantyl group they had a product with better microsomal stability and improved chemical stability. That product was named saxagliptin (Onglyza)(Figure 6).[6] In June 2008 AstraZeneca and Bristol-Myers Squibb submitted a new drug application for Onglyza in the United States and a marketing authorization application in Europe.[13] Approval is still pending.
Denagliptin
Denagliptin (Figure 6) is an advanced compound with a branched side-chain at the P2 position, but also has (4S)-fluoro substitution on the cyanopyrrolidine ring.[6] It is a well-known DPP-4 inhibitor developed by GlaxoSmithKline (GSK). Biological evaluations have shown that the S-configuration of the amino acid portion is essential for the inhibitory activity since the R-configuration showed reluctantly inhibition. These findings will be useful in future designs and synthesis of DPP-4 inhibitors.[14] GSK suspended phase-III clinical trials in October 2008.[15]
Azetidine based compounds
Informations for this group of inhibitors are quite restircted. Azetidine-based DPP-4 inhibitors can roughly be grouped into three main subcategories; 2-cyanoazetidines, 3-fluoroazetidines, and 2-ketoazetidines. The most potent ketoazetidines and cyanoazetidines have large hydrophobic amino acid groups bound to the azetidine nitrogen and are active below 100nM.[16]
Non-substrate-like inhibitors
Non-substrate-like inhibitors do not take after dipeptidic nature of DPP-4 substrates. They are non-covalent inhibitors and usually have an aromatic ring that occupies the S1-pocket, instead of the proline mimetic.[12]
In 1999, Merck started a drug development program on DPP-4 inhibitors. When they started internal screening and medicinal chemistry program, two DPP-4 inhibitors were already in clinical trials, isoleucyl thiazolidide (P32/38) and NVP-DPP728 from Novartis. Merck in-licensed L-threo-isoleucyl thiazolidide and its allo stereoisomer. In animal studies, they found that both isomers had similar affinity for DPP-4, similar in vivo efficacy, similar pharmacokinetic and metabolic profiles. Nevertheless, the allo isomer was 10-fold more toxic. The researchers found out that this difference in toxicity was due to the allo isomer's greater inhibition of DPP-8 and DPP-9 but not because of selective DPP-4 inhibition. More research also supported that DPP-4 inhibition would not cause compromised immune function. Once this link between affinity for DPP-8/DPP-9 and toxicity was discovered, Merck decided on identifying an inhibitor with more than a thousandfold affinity for DPP-4 over the other dipeptidases. For this purpose, they used positional scanning libraries. From scanning these libraries, the researchers discovered that both DPP-4 and DPP-8 showed a strong preference for breaking down peptides with a proline at the P1 position but they found a great difference at the P2 site; i.e., they found that acidic functionality at the P2 position could provide a greater affinity for DPP-4 over DPP-8. Merck kept up doing even more research and screening. They stopped working on compounds from the α-amino acid series related to isoleucyl thiazolidide due to lack of selectivity but instead they discovered a very selective β-amino acid piperazine series through SAR studies on two screening leads. When trying to stabilize the piperazine moiety, a group of bicyclic derivatives were made, which led to the identification of a potent and selective triazolopiperazine series. Most of these analogs showed excellent pharmacokinetic properties in preclinical species. Optimization of these compounds finally led to the discovery of sitagliptin.[17]
Sitagliptin
 Fig.7: The structure of sitagliptin
Fig.7: The structure of sitagliptinSitagliptin (Januvia) has a novel structure with β-amino amide derivatives (Figure 7). Since sitagliptin has shown excellent selectivity and in vivo efficacy it urged researches to inspect the new structure of DPP-4 inhibitors with appended β-amino acid moiety. Further studies are being developed to optimize these compounds for the treatment of diabetes.[4] In October 2006 Sitagliptin became the first DPP-4 inhibitor that got FDA approval for the treatment of type 2 diabetes.[18] Crystallographic structure of sitagliptin along with molecular modeling has been used to continue the search for structurally diverse inhibitors. A new potent, selective and orally bioavailable DPP-4 inhibitor was discovered by replacing the central cyclohexylamine in sitagliptin with 3-aminopiperidine. A 2-pyridyl substitution was the initial SAR breakthrough since that group plays a significant role in potency and selectivity for DPP-4.[2]
It has been shown with an X-ray crystallography how sitagliptin binds to the DPP-4 complex:[12]
1. The trifluorophenyl group occupies the S1-pocket
2. The trifluoromethyl group interacts with the side chains of residues Arg358 and Ser209.
3. The amino group forms a salt bridge with Tyr662 and the carboxylated groups of the two glutamate residues, Glu205 and Glu206.
4. The triazolopiperazine group collides with the phenyl group of residue Phe357
 Fig.8: The structure of ABT-341
Fig.8: The structure of ABT-341Constrained phenylethylamine compounds
Researchers at Abbott Laboratories identified three novel series of DPP-4 inhibitors using HTS. After more research and optimization ABT-341 was discovered (Figure 8). It is a potent and selective DPP-4 inhibitor with a 2D-structure very similar to sitagliptin. However, the 3D-structure is quite different. ABT-341 also has a trifluorophenyl group that occupies the S1-pocket and the free amino group, but the two carbonyl groups are orientated 180° away from each other. ABT-341 is also believed to interact with the Tyr547, probably because of steric hindrance between the cyclohexenyl ring and the tyrosine side-chain.[12]
Pyrrolidine compounds
The pyrrolidine type of DPP-4 inhibitors was first discovered after HTS.[19] Research showed that the pyrrolidine rings were the part of the compounds that fit into the binding site. Further development has led to fluoro substituted pyrrolidines that show superior activity, as well as pyrrolidines with fused cyclopropylrings that are highly active.[20]
Xanthine-based compounds
This is a different class of inhibitors that were identified with HTS.[12] Aromatic heterocyclic-based DPP-4 inhibitors have gained increased attention recently. The first patents describing xanthines (Figure 10) as DPP-4 inhibitors came from Boehringer-Ingelheim(BI) and Novo Nordisk.[21] When xanthine based DPP-4 inhibitors are compared with sitagliptin and vildagliptin it has shown a superior profile. Xanthines are believed to have higher potency, longer-lasting inhibition and longer-lasting improvement of glucose tolerance.[22]
Alogliptin
 Fig.9: Quinazolinone structure and alogliptin
Fig.9: Quinazolinone structure and alogliptinAlogliptin (Figure 9) is a novel DPP-4 inhibitor developed by the Takeda Pharmaceutical Company.[22] Researchers hypothesized that a quinazolinone based structure (Figure 9) would have the necessary groups to interact with the active site on the DPP-4 complex[disambiguation needed
]. Quinazolinone based compounds interacted effectively with the DPP-4 complex, but suffered from low metabolic half-life. It was found that when replacing the quinazolinone with a pyrimidinedione, the metabolic stability was increased and the result was a potent, selective, bioavailable DPP-4 inhibitor named alogliptin. The quinazoline based compounds showed potent inhibition and excellent selectivity over related protease, DPP-8. However, short metabolic half-life due to oxidation of the A-ring phenyl group was problematic. At first, the researchers tried to make a fluorinated derivative. The derivative showed improved metabolic stability and excellent inhibition of the DPP-4 enzyme. However, it was also found to inhibit CYP 450 3A4 and block the hERG channel. The solution to this problem was to replace the quinazolinone with other heterocycles, but the quinazolinone could be replaced without any loss of DPP-4 inhibition. Alogliptin was discovered when quinazolinone was replaced with a pyrimidinedione. Alogliptin has shown excellent inhibition of DPP-4 and extraordinary selectivity, greater than 10.000 fold over the closely related serine proteases DPP-8 and DPP-9. Also, it does not inhibit the CYP 450 enzymes nor block the hERG channel at concentration up to 30µM. Based on this data, alogliptin was chosen for preclinical evaluation.[23] In January 2007 alogliptin was undergoing the phase III clinical trial and in October 2008 it was being reviewed by the U.S. Food and Drug Administration.[24]Linagliptin
 Fig.10: The structure of xanthine type inhibitors and linagliptin (known as BI-1356 in clinical trials)
Fig.10: The structure of xanthine type inhibitors and linagliptin (known as BI-1356 in clinical trials)Researchers at BI discovered that using a buty-2-nyl group resulted in a potent candidate, called BI-1356 (Figure 10). In 2008 BI-1356 was undergoing phase III clinical trials; it was released as linagliptin in May 2011. X-ray crystallography has shown that that xanthine type binds the DPP-4 complex in a different way than other inhibitors:[12]
1. The amino group also interacts with the Glu205, Glu206 and Tyr662
2. The buty-2-nyl group occupies the S1-pocket
3. The uracil group undergoes a π-stacking interaction with the Tyr547 residue
4. The quinazoline group undergoes a π-stacking interaction with the Trp629 residue
Pharmacology
Comparative pharmacology of sitagliptin and vildagliptin.[18] Drug Absorption Bioavailability (%) IC50 (nM) Mean volume of distribution (L) Protein binding (%) Half-life (hours,100 mg dose) Metabolism Excretion Sitagliptin Rapidly absorbed with peak concentration at 1–4 hours 87 18 198 38 12.4 Small fraction undergoes hepatic metabolism via CYP 450 3A4 and 2C8 Excreted in an unchanged form in the urine (79%) Vildagliptin Rapidly absorbed with peak concentration at 1–2 hours 85 3 70.5 9.3 1.68 (once a day) and 2.54 (twice a day) Hydrolysis resulting in a pharmacologically inactive metabolite. A fraction (22%) is also excreted unchanged by the kidneys Excretion of the metabolite is carried out through urine (85%) and feces (15%) The pharmacokinetic properties of sitagliptin and vildagliptin appear unaffected by age, sex or BMI.[18] Clinical researches have shown that sitagliptin and vildagliptin do not have the side effects that tend to follow type 2 diabetes treatment, e.g. weight gain and hyperglycemia, but however, other side effects have been observed, including upper respiratory tract infections, sore throat and diarrhea.[5]
See also
- Dipeptidyl peptidase-4
- Dipeptidyl peptidase-4 inhibitors
- Vildagliptin
- Sitagliptin
- Saxagliptin
- Berberine
References
- ^ a b Green, Brian; Flatt, Peter; Bailey, Clifford (December 2006), "Dipeptidyl peptidase IV (DPP IV) inhibitors: a newly emerging drug class for the treatment of type 2 diabetes", Diabetes and vascular disease research 3 (3): 159–165, doi:10.3132/dvdr.2006.024, PMID 17160910, http://www.dvdres.com/pdf/2552.pdf[dead link]
- ^ a b c d Sebokova, Elena; Christ, Andreas; Boehringer, Markus; Mizrahi, Jacques (May 2006), "Dipeptidyl peptidase IV inhibitors: The next generation of new promising therapies for the management of type 2 diabetes", Current Topics in Medicinal Chemistry 7 (6): 547–555, doi:10.2174/156802607780091019, PMID 17352676, http://www.ingentaconnect.com/content/ben/ctmc/2007/00000007/00000006/art00002
- ^ a b c Wiedeman, Paul (May 2007), "DPP-IV Inhibition: Promising Therapy for the Treatment of Type 2 Diabetes", Progress in Medical Chemistry 45: 71–73, ISBN 9780444528087, http://books.google.com/?id=-WxwxEIzB3EC&pg=PA63&dq=dppiv+inhibition+wiedeman
- ^ a b Ahn, Sung Soo; Shin, Mi Sik; Jun, Mi Ae; Jung, Sun Ho; Kang, Seung Kyu; Kim, Kwang Rok; Rhee, Sang Dal; Kang, Nam Sook et al. (February 2007), "Synthesis, biological evaluation and structural determination of β-aminoacyl-containing cyclic hydrazine derivatives as dipeptidyl peptidase IV (DPP-IV) inhibitors", Bioorganic & Medicinal Chemistry Letters 17 (9): 2622–2628, doi:10.1016/j.bmcl.2007.01.111, PMID 17331715, http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6VKY-4R7NPSY-1&_user=713789&_rdoc=1&_fmt=&_orig=search&_sort=d&view=c&_version=1&_urlVersion=0&_userid=713789&md5=914f71fb6b477e3998e33cf49c91b6d9
- ^ a b c Lu, Su-Ying; Tsai, Keng-Chang; Chiang, Yi-Kun; Jiaang, Weir-Torn; Wu, Ssu-Hui; Mahindroo, Neeraj; Chien, Chia-Hui; Lee, Shiow-Ju et al. (January 2008), "A three-dimensional pharmacophore model for dipeptidyl peptidase IV inhibitors", European Journal of Medicinal Chemistry 43 (8): 1603–1611, doi:10.1016/j.ejmech.2007.11.014, PMID 18207285, http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6VKY-4R7NPSY-1&_user=713789&_rdoc=1&_fmt=&_orig=search&_sort=d&view=c&_version=1&_urlVersion=0&_userid=713789&md5=914f71fb6b477e3998e33cf49c91b6d9
- ^ a b c d e f g h Peters, Jens-Uwe (March 2007), "11 years of Cyanopyrrolidines as DPP-IV Inhibitors", Current Topics in Medicinal Chemistry 7 (6): 579–595, doi:10.2174/156802607780091000, PMID 17352679, http://www.ingentaconnect.com/content/ben/ctmc/2007/00000007/00000006/art00005
- ^ Cox, Scott; Harper, Bart; Mastracchio, Anthony; Leiting, Barbara; Roy, Ranabir; Reshma, Patel; Wu, Joseph; Lyons, Kathryn et al. (June 2007), "Discovery of 3-aminopiperidines as potent, selective, and orally bioavailable dipeptidyl peptidase IV inhibitors", Bioorganic & Medicinal Chemistry 17 (16): 4579–4583, doi:10.1016/j.bmcl.2007.05.087, PMID 17562364, http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6TF9-4NWNCPF-1&_user=713789&_rdoc=1&_fmt=&_orig=search&_sort=d&view=c&_acct=C000039858&_version=1&_urlVersion=0&_userid=713789&md5=20851c5fcfe5679554793cf12461ba7d
- ^ a b Idris, Iskandar; Donnely, Richard (January), "Dipeptidyl peptidase-IV inhibitors: a major new class of oral antidiabetic drug", Diabetes, Obesity and Metabolism 9 (2): 153–165, doi:10.1111/j.1463-1326.2007.00705.x, PMID 17300591, http://pt.wkhealth.com/pt/re/dome/abstract.00127973-200703000-00002.htm;jsessionid=L8HQg0MX3QSvk2bPvN8BgynJ8zZ2r1JXrvHxmYpQRH1ntqR1F2jR!949623904!181195628!8091!-1
- ^ a b Kuhn, Bernd; Hennig, Michael; Mattei, Patrizio (March 2007), "Molecular Recognition of Ligands in Dipeptidyl Peptidase IV", Current Topics in Medicinal Chemistry 7 (6): 609–619, doi:10.2174/156802607780091064, PMID 17352681, http://www.ingentaconnect.com/content/ben/ctmc/2007/00000007/00000006/art00007
- ^ Veken, Pieter Van der; Haemers, Achiel; Augustyns, Koen (May 2007), "Prolyl peptidase related to dipeptidyl peptidase IV: Potential of specific inhibitors in drug discovery", Current Topics in Medicinal Chemistry 7 (6): 621–635, doi:10.2174/156802607780091046, PMID 17352682, http://www.ingentaconnect.com/content/ben/ctmc/2007/00000007/00000006/art00008
- ^ a b Simpkins, Lawrence; Bolton, Scott; Pi, Zulan; Scutton, James; Kwon, Chet; Magnin, David; Augeri, David; Gungor, Timur et al. (September 2007), "Potent non-nitrile dipeptidic dipeptidyl peptidase IV inhibitors", Bioorganic & Medicinal Chemistry Letters 17 (23): 6476–6480, doi:10.1016/j.bmcl.2007.09.090, PMID 17937986, http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6TF9-4PT1SC6-1&_user=713789&_rdoc=1&_fmt=&_orig=search&_sort=d&view=c&_version=1&_urlVersion=0&_userid=713789&md5=417ae5d6a31a5ab0d6dd3e9d639f55e2
- ^ a b c d e f g Pei, Zhonghua (March 2007), "From the bench to the bedside: Dipeptidyl peptidase IV inhibitors, a new class of oral antihyperglycemic agents", Current Opinion in Drug Discovery & Development 11 (4): 515–532, http://www.biomedcentral.com/render/render.asp?arx_id=cd-921157&type=pdf&access=denied
- ^ , http://www.astrazeneca-us.com/search/?itemId=3310018
- ^ Deng, Guanghui; Ye, Deju; Li, Yuanyuan; He, Lingyan; Zhou, Yu; Wang, Jiang; Li, Jia; Jiang, Hualiang et al. (September 2008), "Synthesis of (S)-, (R)-, and (rac)-2-amino-3,3-bis(4-fluorophenyl)propanoic acids and an evaluation of the DPP-IV inhibitory activity of Denagliptin diastereomers", Tetrahedron 64: 10512–10516, doi:10.1016/j.tet.2008.08.097, http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6THR-4TCR1KY-1&_user=713789&_rdoc=1&_fmt=&_orig=search&_sort=d&view=c&_version=1&_urlVersion=0&_userid=713789&md5=c7c0693b91083f2be5e1d9c4052a8449
- ^ , http://www.gsk-clinicalstudyregister.com/protocol_detail.jsp?protocolId=DPB107246&studyId=20659&compound=Denagliptin
- ^ Ferraris, Dana; Belyakov, Sergei; Li, Weixing; Oliver, Eddie; Ko, Yao-Sen; Calvin, David; Lautar, Susan; Thomas, Bert et al. (March 2007), "Azetidine-Based Inhibitors of Dipeptidyl Peptidase IV (DPP IV)", Current Topics in Medicinal Chemistry 7 (6): 597–608, doi:10.2174/156802607780090993, PMID 17352680, http://www.ingentaconnect.com/content/ben/ctmc/2007/00000007/00000006/art00006
- ^ Thornberry, Nancy A.; Weber, Ann E. (March 2007), "Discovery of JANUVIATM (Sitagliptin), a Selective Dipeptidyl Peptidase IV Inhibitor for the Treatment of Type2 Diabetes", Current Topics in Medicinal Chemistry 7 (6): 557–568, doi:10.2174/156802607780091028, PMID 17352677, http://www.ingentaconnect.com/content/ben/ctmc/2007/00000007/00000006/art00003
- ^ a b c White, John (2008), "Dipeptidyl Peptidase-IV Inhibitors: Pharmacological Profile and Clinical Use", Clinical Diabetes 26 (2): 53–57, doi:10.2337/diaclin.26.2.53, http://clinical.diabetesjournals.org/cgi/reprint/26/2/53
- ^ Wright, Cox; Ammirati, Mark; Andrews, Kim; Brodeur, Anne; Danley, Dennis; Doran, Shawn; Lillquist, Jay; Liu, Shenping et al. (August 2007), "(3R,4S)-4-(2,4,5-Trifluorophenyl)-pyrrolidin-3-ylamine inhibitors of dipeptidyl peptidase IV: Synthesis, in vitro, in vivo, and X-ray crystallographic characterization", Bioorganic & Medicinal Chemistry Letters 17 (20): 5638–5642, doi:10.1016/j.bmcl.2007.07.081, PMID 17822893, http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6TF9-4PGGP17-3&_user=713789&_rdoc=1&_fmt=&_orig=search&_sort=d&view=c&_version=1&_urlVersion=0&_userid=713789&md5=f71c1030985c0de57f8372a1c16794b7
- ^ Hulin, Bernard; Cabral, Shawn; Lopaze, Michael; VanVolkenburg, Maria; Andrews, Kim; Parker, Janice (August 2005), "New fluorinated pyrrolidine and azetidine amides as dipeptidyl peptidase IV inhibitors", Bioorganic & Medicinal Chemistry Letters 17: 4770–4773, http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6TF9-4GXVG46-8&_user=713789&_rdoc=1&_fmt=&_orig=search&_sort=d&view=c&_acct=C000039858&_version=1&_urlVersion=0&_userid=713789&md5=ae8f3ea9b4d91dc112dfba5acf970d62
- ^ Szczepankiewicz, Bruce G.; Kurukulasuriya, Ravi (March 2007), "Aromatic Heterocycle-Based DPP-IV Inhibitors: Xanthines and Related Structural Types", Current Topics in Medicinal Chemistry 7 (6): 569–578, doi:10.2174/156802607780091073, PMID 17352678, http://www.ingentaconnect.com/content/ben/ctmc/2007/00000007/00000006/art00004
- ^ a b Ferrannini, E.; Skrha, J.; Li, Eizirik; D.L., Gale; E., Jörgens; V. (2007), "Minutes of the 42nd general assembly of the European Association for the study of diabetes", Diabetologia 50: 362–363, http://proquest.umi.com/pqdlink?index=0&did=1325012151&SrchMode=1&sid=1&Fmt=6&VInst=PROD&VType=PQD&RQT=309&VName=PQD&TS=1226402456&clientId=58032
- ^ Feng, Jun; Zhang, Zhiyuan; Wallace, Michael; Stafford, Jeffrey; Kaldor, Stephen; Kassel, Daniel; Navre, Marc; Shi, Lihong et al. (January 2007), "Discovery of alogliptin: A potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV", Journal of Medicinal Chemistry 50 (10): 2297–2300, doi:10.1021/jm070104l, PMID 17441705, http://pubs.acs.org/cgi-bin/abstract.cgi/jmcmar/2007/50/i10/abs/jm070104l.html
- ^ , http://www.takeda.com/press/article_31125.html
Categories:








Wikimedia Foundation. 2010.