- Galactose-1-phosphate uridylyltransferase deficiency
-
Galactose-1-phosphate uridylyltransferase deficiency Classification and external resources 
GalactoseICD-10 E74.2 ICD-9 271.1 OMIM 230400 DiseasesDB 5056 eMedicine ped/818 MeSH D005693 Galactose-1-phosphate uridylyltransferase deficiency, also called galactosemia type 1, classic galactosemia or GALT deficiency, is the most common type of galactosemia, an inborn error of galactose metabolism, caused by a deficiency of the enzyme galactose-1-phosphate uridylyltransferase.[1] It is an autosomal recessive metabolic disorder caused by a deficiency of the enzyme galactose-1-phosphate uridylyltransferase.
Contents
Cause
After the ingestion of lactose, the enzyme lactase hydrolyzes the sugar into glucose and galactose. Galactose is converted to galactose 1-phosphate (Gal-1-P) by the enzyme galactokinase. Gal-1-P is converted to uridine diphosphate (UDP) galactose by the enzyme galactose-1-phosphate uridylyltransferase, with UDP-glucose acting as a UDP donor. UDP-galactose can then be converted to lactose, by the enzyme lactose synthase, or to UDP-glucose by UDP galactose epimerase. In classic galactosemia, galactose-1-phosphate uridylyltransferase activity is reduced or absent, leading to an accumulation of galactose and Gal-1-P.[2]
Symptoms & Diagnosis
In untreated children, the accumulation of metabolites due to the deficient activity of galactose 1-phosphate uridylyltransferase can lead to feeding problems, failure to thrive, liver damage, bleeding and infections. The first presenting symptom in an infant is often prolonged jaundice. Without intervention in the form of galactose restriction, infants can develop hyperammonemia and sepsis, possibly leading to shock. The accumulation of Gal-1-P can lead to cataracts which are similar to those seen in galactokinase deficiency.[3] Long-term consequences of continued galactose intake can include developmental delay, verbal dyspraxia and motor abnormalities. Galactosemic females frequently suffer from ovarian failure.[3]
In most regions, galactosemia is diagnosed as a result of newborn screening, most commonly by determining the concentration of galactose in a dried blood spot. Some regions will perform a second-tier test of GALT enzyme activity on samples with elevated galactose, while others will immediately initiate confirmatory testing. While awaiting confirmatory testing for classic galactosemia, the infant is typically fed a soy-based formula, as human and cow milk contains galactose.[4] Confirmatory testing would include measurement of enzyme activity in red blood cells, determination of Gal-1-P levels in the blood, and genetic testing.
Genetics
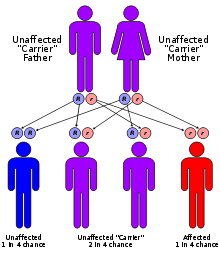 Galactose-1-phosphate uridylyltransferase galactosemia has an autosomal recessive pattern of inheritance.
Galactose-1-phosphate uridylyltransferase galactosemia has an autosomal recessive pattern of inheritance.
There are several variants in the GALT gene, which have different levels of residual enzyme activity. A patient homozygous for the one of the severe mutations in the GALT gene (commonly referred to as G/G) will typically have less than 55 of the enzyme activity expected in an unaffected patient.[3] Duarte galactosemia is caused by mutations that produce an unstable form of the GALT enzyme, with reduced promoter expression. Patients who are homozygous for Duarte mutations (D/D) will have reduced levels of enzyme activity compared to normal controls, but can often maintain a normal diet. Compound heterozygotes (D/G) will often be detected by newborn screening, and treatment is based on the extent of residual enzyme activity.[3]
References
- ^ Online 'Mendelian Inheritance in Man' (OMIM) 230400
- ^ Salway, J.G. (2004). Metabolism at a Glance (3rd ed.). Malden, MA: Blackwell. p. 102.
- ^ a b c d "Galactosemia". U.S. National Library of Medicine. http://www.ncbi.nlm.nih.gov/books/NBK1518/. Retrieved 2011-10-16.
- ^ "Newborn Screening ACT Sheet [Absent/Reduced Galactose-1-Phosphate Uridyltransferase (GALT) Classical Galactosemia"]. American College of Medical Genetics. http://www.acmg.net/StaticContent/ACT/Galactose+GALT.pdf. Retrieved 2011-11-05.
Inborn error of carbohydrate metabolism: monosaccharide metabolism disorders (including glycogen storage diseases) (E73–E74, 271) Sucrose, transport
(extracellular)Disaccharide catabolismMonosaccharide transportHexose → glucose Monosaccharide catabolismGlucose ⇄ glycogen Glucose ⇄ CAC Pentose phosphate pathway Other Categories:- Inborn errors of carbohydrate metabolism
- Autosomal recessive disorders
- Genetic disorder stubs


Wikimedia Foundation. 2010.