- Atherosclerosis
-
For the journal, see Atherosclerosis (journal).
Atherosclerosis Classification and external resources 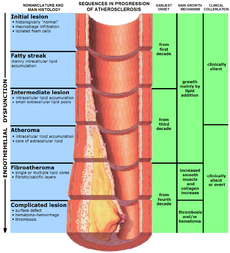
The progression of atherosclerosis (size exaggerated; see text)ICD-10 I70 ICD-9 440, 414.0 DiseasesDB 1039 MedlinePlus 000171 eMedicine med/182 MeSH D050197 Atherosclerosis (also known as arteriosclerotic vascular disease or ASVD) is a condition in which an artery wall thickens as a result of the accumulation of fatty materials such as cholesterol. It is a syndrome affecting arterial blood vessels, a chronic inflammatory response in the walls of arteries, caused largely by the accumulation of macrophage white blood cells and promoted by low-density lipoproteins (plasma proteins that carry cholesterol and triglycerides) without adequate removal of fats and cholesterol from the macrophages by functional high density lipoproteins (HDL), (see apoA-1 Milano). It is commonly referred to as a hardening or furring of the arteries. It is caused by the formation of multiple plaques within the arteries.[1]
The atheromatous plaque is divided into three distinct components:
- The atheroma ("lump of gruel," from ἀθήρα, athera, gruel in Greek), which is the nodular accumulation of a soft, flaky, yellowish material at the center of large plaques, composed of macrophages nearest the lumen of the artery
- Underlying areas of cholesterol crystals
- Calcification at the outer base of older/more advanced lesions.
The following terms are similar, yet distinct, in both spelling and meaning, and can be easily confused: arteriosclerosis, arteriolosclerosis, and atherosclerosis. Arteriosclerosis is a general term describing any hardening (and loss of elasticity) of medium or large arteries (from the Greek arteria, meaning artery, and sclerosis, meaning hardening); arteriolosclerosis is any hardening (and loss of elasticity) of arterioles (small arteries); atherosclerosis is a hardening of an artery specifically due to an atheromatous plaque. The term atherogenic is used for substances or processes that cause atherosclerosis.
Atherosclerosis is a chronic disease that remains asymptomatic for decades.[2] Atherosclerotic lesions, or atherosclerotic plaques are separated into two broad categories: Stable and unstable (also called vulnerable).[3] The pathobiology of atherosclerotic lesions is very complicated but generally, stable atherosclerotic plaques, which tend to be asymptomatic, are rich in extracellular matrix and smooth muscle cells, while, unstable plaques are rich in macrophages and foam cells and the extracellular matrix separating the lesion from the arterial lumen (also known as the fibrous cap) is usually weak and prone to rupture.[4] Ruptures of the fibrous cap expose thrombogenic material, such as collagen [5] to the circulation and eventually induce thrombus formation in the lumen. Upon formation, intraluminal thrombi can occlude arteries outright (i.e. coronary occlusion), but more often they detach, move into the circulation and eventually occlude smaller downstream branches causing thromboembolism (i.e. Stroke is often caused by thrombus formation in the carotid arteries). Apart from thromboembolism, chronically expanding atherosclerotic lesions can cause complete closure of the lumen. Interestingly, chronically expanding lesions are often asymptomatic until lumen stenosis is so severe that blood supply to downstream tissue(s) is insufficient resulting in ischemia.
These complications of advanced atherosclerosis are chronic, slowly progressive and cumulative. Most commonly, soft plaque suddenly ruptures (see vulnerable plaque), causing the formation of a thrombus that will rapidly slow or stop blood flow, leading to death of the tissues fed by the artery in approximately 5 minutes. This catastrophic event is called an infarction. One of the most common recognized scenarios is called coronary thrombosis of a coronary artery, causing myocardial infarction (a heart attack). The same process in an artery to the brain is commonly called stroke. Another common scenario in very advanced disease is claudication from insufficient blood supply to the legs, typically caused by a combination of both stenosis and aneurysmal segments narrowed with clots.
Atherosclerosis can occur body-wide, in the arteries to the brain, intestines, kidneys, legs, etc. with many infarctions involving only very small amounts of tissue. These are termed "clinically silent" because the person having the infarction does not notice the problem and does not seek medical help, or when they do, physicians do not recognize what has happened.
Contents
Signs and symptoms
Atherosclerosis typically begins in early adolescence, and is usually found in most major arteries, yet is asymptomatic and not detected by most diagnostic methods during life. Atheroma in arm, or more often in leg arteries, which produces decreased blood flow is called peripheral artery occlusive disease (PAOD).
According to United States data for the year 2004, for about 65% of men and 47% of women, the first symptom of atherosclerotic cardiovascular disease is heart attack or sudden cardiac death (death within one hour of onset of the symptom).[citation needed]
Most artery flow disrupting events occur at locations with less than 50% lumen narrowing (~20% stenosis is average). The illustration above, like most illustrations of arterial disease, overemphasizes lumen narrowing, as opposed to compensatory external diameter enlargement (at least within smaller arteries, e.g., heart arteries) typical of the atherosclerosis process as it progresses (see Glagov[6] or the ASTEROID trial[7]). The relative geometry error within the illustration is common to many older illustrations, an error slowly being more commonly recognized within the last decade.
Cardiac stress testing, traditionally the most commonly performed non-invasive testing method for blood flow limitations, in general, detects only lumen narrowing of ~75% or greater, although some physicians claim that nuclear stress methods can detect as little as 50%.
Causes
The main cause of atherosclerosis is yet unknown, but is hypothesized to fundamentally be initiated by inflammatory processes in the cell wall in response to retained low-density lipoprotein (LDL) molecules.[8] Once inside the vessel wall, LDL molecules become susceptible to oxidation by free radicals,[9] and become toxic to the cells. The damage caused by the oxidized LDL molecules triggers a cascade of immune responses which over time can produce an atheroma. The LDL molecule is globular shaped with a hollow core to carry cholesterol throughout the body.
The body's immune system responds to the damage to the artery wall caused by oxidized LDL by sending specialized white blood cells (macrophages and T-lymphocytes) to absorb the oxidized-LDL forming specialized foam cells. These white blood cells are not able to process the oxidized-LDL, and ultimately grow then rupture, depositing a greater amount of oxidized cholesterol into the artery wall. This triggers more white blood cells, continuing the cycle.
Eventually, the artery becomes inflamed. The cholesterol plaque causes the muscle cells to enlarge and form a hard cover over the affected area. This hard cover is what causes a narrowing of the artery, reduces the blood flow and increases blood pressure.
Some researchers believe that atherosclerosis may be caused by an infection of the vascular smooth muscle cells; chickens, for example, develop atherosclerosis when infected with the Marek's disease herpesvirus.[10] Herpesvirus infection of arterial smooth muscle cells has been shown to cause cholesteryl ester (CE) accumulation.[11] Cholesteryl ester accumulation is associated with atherosclerosis.
Also, cytomegalovirus (CMV) infection is associated with cardiovascular diseases.[12]
Linus Pauling's and Matthias Rath's extended theory [13] states that deaths from scurvy in humans during the ice age, when vitamin C (an antioxidant) was scarce, selected for individuals who could repair arteries with a layer of cholesterol provided by lipoprotein(a), a lipoprotein found in vitamin C-deficient species (higher primates and guinea pigs). Pauling and Rath hypothesized that, although eventually harmful, lipoprotein deposition on artery walls was beneficial to the human species and a "surrogate for ascorbate" in that it kept individuals alive until access to vitamin C allowed arterial damage to be repaired. Atherosclerosis is thus a vitamin-C-deficiency disease.
Risk factors
Various anatomic, physiological and behavioral risk factors for atherosclerosis are known.[14] These can be divided into various categories: congenital vs acquired, modifiable or not, classical or non-classical. The points labelled '+' in the following list form the core components of metabolic syndrome.
Risks multiply, with two factors increasing the risk of atherosclerosis fourfold.[15] Hyperlipidemia, hypertension and cigarette smoking together increases the risk seven times.[15]
- Modifiable
- Diabetes[15] or Impaired glucose tolerance (IGT) +
- Dyslipoproteinemia[15] (unhealthy patterns of serum proteins carrying fats & cholesterol): +
- High serum concentration of low-density lipoprotein (LDL, "bad if elevated concentrations and small"), and / or very low density lipoprotein (VLDL) particles, i.e., "lipoprotein subclass analysis"
- Low serum concentration of functioning high density lipoprotein (HDL "protective if large and high enough" particles), i.e., "lipoprotein subclass analysis"
- An LDL:HDL ratio greater than 3:1
- Tobacco smoking, increases risk by 200% after several pack years[15]
- Having hypertension +, on its own increasing risk by 60%[15]
- Elevated serum C-reactive protein concentrations[15][16]
- Vitamin B6 deficiency[17][18][19]
- Nonmodifiable
- Advanced age[15]
- Male sex[15]
- Having close relatives who have had some complication of atherosclerosis (e.g. coronary heart disease or stroke)[15]
- Genetic abnormalities,[15] e.g. familial hypercholesterolemia
- Lesser or uncertain
The following factors are of relatively lesser importance, are uncertain or unquantified:
- Obesity[15] (in particular central obesity, also referred to as abdominal or male-type obesity) +
- A sedentary lifestyle[15]
- Hypercoagulability[20][21][22]
- Postmenopausal estrogen deficiency[15]
- High intake of saturated fat (may raise total and LDL cholesterol)[23]
- Intake of trans fat (may raise total and LDL cholesterol while lowering HDL cholesterol)[15][24]
- High carbohydrate intake[15]
- Elevated serum levels of triglycerides +
- Elevated serum levels of homocysteine
- Elevated serum levels of uric acid (also responsible for gout)
- Elevated serum fibrinogen concentrations
- Elevated serum lipoprotein(a) concentrations[15]
- Chronic systemic inflammation as reflected by upper normal WBC concentrations, elevated hs-CRP and many other blood chemistry markers, most only research level at present, not clinically done.[25]
- Stress[15] or symptoms of clinical depression
- Hyperthyroidism (an over-active thyroid)
- Elevated serum insulin levels +[26]
- Short sleep duration[27]
- Chlamydia pneumoniae infection[15]
- Dietary
The relation between dietary fat and atherosclerosis is a contentious field. The USDA, in its food pyramid, promotes a low-fat diet, based largely on its view that fat in the diet is atherogenic. The American Heart Association, the American Diabetes Association and the National Cholesterol Education Program make similar recommendations. In contrast, Prof Walter Willett (Harvard School of Public Health, PI of the second Nurses' Health Study) recommends much higher levels, especially of monounsaturated and polyunsaturated fat.[28] Writing in Science, Gary Taubes detailed that political considerations played into the recommendations of government bodies.[29] These differing views reach a consensus, though, against consumption of trans fats.
The role of dietary oxidized fats / lipid peroxidation (rancid fats) in humans is not clear. Laboratory animals fed rancid fats develop atherosclerosis. Rats fed DHA-containing oils experienced marked disruptions to their antioxidant systems, as well as accumulated significant amounts of phospholipid hydroperoxide in their blood, livers and kidneys.[30] In another study, rabbits fed atherogenic diets containing various oils were found to undergo the greatest amount of oxidative susceptibility of LDL via polyunsaturated oils.[31] In a study involving rabbits fed heated soybean oil, "grossly induced atherosclerosis and marked liver damage were histologically and clinically demonstrated."[32]
Rancid fats and oils taste very bad even in small amounts; people avoid eating them.[33] It is very difficult to measure or estimate the actual human consumption of these substances.[34] In addition, the majority of oils consumed in the United States are refined, bleached, deodorized and degummed by manufacturers. The resultant oils are colorless, odorless, tasteless and have a longer shelf life than their unrefined counterparts.[35] This extensive processing serves to make peroxidated, rancid oils much more elusive to detection via the various human senses than the unprocessed alternatives.
It is necessary to note that highly unsaturated omega-3 rich oil such as fish oil are being sold in pill form so that the taste of oxidized or rancid fat is not apparent. The health food industry dietary supplement are self regulated by the manufacture and outside of FDA regulations.[36] To properly protect unsaturated fats from oxidation, it is best to keep them cool and in oxygen free environments.
Pathophysiology
Atherogenesis is the developmental process of atheromatous plaques. It is characterized by a remodeling of arteries leading to subendothelial accumulation of fatty substances called plaques. The build up of an atheromatous plaque is a slow process, developed over a period of several years through a complex series of cellular events occurring within the arterial wall, and in response to a variety of local vascular circulating factors. One recent theory suggests that, for unknown reasons, leukocytes, such as monocytes or basophils, begin to attack the endothelium of the artery lumen in cardiac muscle. The ensuing inflammation leads to formation of atheromatous plaques in the arterial tunica intima, a region of the vessel wall located between the endothelium and the tunica media. The bulk of these lesions is made of excess fat, collagen, and elastin. At first, as the plaques grow, only wall thickening occurs without any narrowing. Stenosis is a late event, which may never occur and is often the result of repeated plaque rupture and healing responses, not just the atherosclerotic process by itself.
Cellular
 Micrograph of an artery that supplies the heart with significant atherosclerosis and marked luminal narrowing. Tissue has been stained using Masson's trichrome.
Micrograph of an artery that supplies the heart with significant atherosclerosis and marked luminal narrowing. Tissue has been stained using Masson's trichrome.
Early atherogenesis is characterized by the adherence of blood circulating monocytes to the vascular bed lining, the endothelium, followed by their migration to the sub-endothelial space, and further activation into monocyte-derived macrophages.[37] The primary documented driver of this process is oxidized Lipoprotein particles within the wall, beneath the endothelial cells, though upper normal or elevated concentrations of blood glucose also plays a major role and not all factors are fully understood. Fatty streaks may appear and disappear.
Low Density Lipoprotein particles in blood plasma, when they invade the endothelium and become oxidized creates a risk for cardiovascular disease. A complex set of biochemical reactions regulates the oxidation of LDL, chiefly stimulated by presence of enzymes, e.g. Lp-LpA2 and free radicals in the endothelium or blood vessel lining.
The initial damage to the blood vessel wall results in an inflammatory response. Monocytes (a type of white blood cell) enter the artery wall from the bloodstream, with platelets adhering to the area of insult. This may be promoted by redox signaling induction of factors such as VCAM-1, which recruit circulating monocytes. The monocytes differentiate into macrophages, which ingest oxidized LDL, slowly turning into large "foam cells" – so-described because of their changed appearance resulting from the numerous internal cytoplasmic vesicles and resulting high lipid content. Under the microscope, the lesion now appears as a fatty streak. Foam cells eventually die, and further propagate the inflammatory process. There is also smooth muscle proliferation and migration from tunica media to intima responding to cytokines secreted by damaged endothelial cells. This would cause the formation of a fibrous capsule covering the fatty streak.
Calcification and lipids
Intracellular microcalcifications form within vascular smooth muscle cells of the surrounding muscular layer, specifically in the muscle cells adjacent to the atheromas. In time, as cells die, this leads to extracellular calcium deposits between the muscular wall and outer portion of the atheromatous plaques. A similar form of an intramural calcification, presenting the picture of an early phase of arteriosclerosis, appears to be induced by a number of drugs that have an antiproliferative mechanism of action (Rainer Liedtke 2008).
Cholesterol is delivered into the vessel wall by cholesterol-containing low-density lipoprotein (LDL) particles. To attract and stimulate macrophages, the cholesterol must be released from the LDL particles and oxidized, a key step in the ongoing inflammatory process. The process is worsened if there is insufficient high-density lipoprotein (HDL), the lipoprotein particle that removes cholesterol from tissues and carries it back to the liver.
The foam cells and platelets encourage the migration and proliferation of smooth muscle cells, which in turn ingest lipids, become replaced by collagen and transform into foam cells themselves. A protective fibrous cap normally forms between the fatty deposits and the artery lining (the intima).
These capped fatty deposits (now called 'atheromas') produce enzymes that cause the artery to enlarge over time. As long as the artery enlarges sufficiently to compensate for the extra thickness of the atheroma, then no narrowing ("stenosis") of the opening ("lumen") occurs. The artery becomes expanded with an egg-shaped cross-section, still with a circular opening. If the enlargement is beyond proportion to the atheroma thickness, then an aneurysm is created.[6]
Visible features
Although arteries are not typically studied microscopically, two plaque types can be distinguished:[38]
- The fibro-lipid (fibro-fatty) plaque is characterized by an accumulation of lipid-laden cells underneath the intima of the arteries, typically without narrowing the lumen due to compensatory expansion of the bounding muscular layer of the artery wall. Beneath the endothelium there is a "fibrous cap" covering the atheromatous "core" of the plaque. The core consists of lipid-laden cells (macrophages and smooth muscle cells) with elevated tissue cholesterol and cholesterol ester content, fibrin, proteoglycans, collagen, elastin, and cellular debris. In advanced plaques, the central core of the plaque usually contains extracellular cholesterol deposits (released from dead cells), which form areas of cholesterol crystals with empty, needle-like clefts. At the periphery of the plaque are younger "foamy" cells and capillaries. These plaques usually produce the most damage to the individual when they rupture.
- The fibrous plaque is also localized under the intima, within the wall of the artery resulting in thickening and expansion of the wall and, sometimes, spotty localized narrowing of the lumen with some atrophy of the muscular layer. The fibrous plaque contains collagen fibers (eosinophilic), precipitates of calcium (hematoxylinophilic) and, rarely, lipid-laden cells.
In effect, the muscular portion of the artery wall forms small aneurysms just large enough to hold the atheroma that are present. The muscular portion of artery walls usually remain strong, even after they have remodeled to compensate for the atheromatous plaques.
However, atheromas within the vessel wall are soft and fragile with little elasticity. Arteries constantly expand and contract with each heartbeat, i.e., the pulse. In addition, the calcification deposits between the outer portion of the atheroma and the muscular wall, as they progress, lead to a loss of elasticity and stiffening of the artery as a whole.
The calcification deposits, after they have become sufficiently advanced, are partially visible on coronary artery computed tomography or electron beam tomography (EBT) as rings of increased radiographic density, forming halos around the outer edges of the atheromatous plaques, within the artery wall. On CT, >130 units on the Hounsfield scale (some argue for 90 units) has been the radiographic density usually accepted as clearly representing tissue calcification within arteries. These deposits demonstrate unequivocal evidence of the disease, relatively advanced, even though the lumen of the artery is often still normal by angiographic or intravascular ultrasound.
In days gone by the lateral chest x-ray (demonstrating greater opacity in the aortic arch and descending aorta than the thoracic spine) gave an indication to the degree of calcified plaque burden a patient had. This has been known as Piper's sign and can often be seen in elderly persons particularly those with concomitant osteoporosis.
Rupture and stenosis
Although the disease process tends to be slowly progressive over decades, it usually remains asymptomatic until an atheroma ulcerates, which leads to immediate blood clotting at the site of atheroma ulcer. This triggers a cascade of events that leads to clot enlargement, which may quickly obstruct the flow of blood. A complete blockage leads to ischemia of the myocardial (heart) muscle and damage. This process is the myocardial infarction or "heart attack".
If the heart attack is not fatal, fibrous organization of the clot within the lumen ensues, covering the rupture but also producing stenosis or closure of the lumen, or over time and after repeated ruptures, resulting in a persistent, usually localized stenosis or blockage of the artery lumen. Stenoses can be slowly progressive, whereas plaque ulceration is a sudden event that occurs specifically in atheromas with thinner/weaker fibrous caps that have become "unstable".
Repeated plaque ruptures, ones not resulting in total lumen closure, combined with the clot patch over the rupture and healing response to stabilize the clot, is the process that produces most stenoses over time. The stenotic areas tend to become more stable, despite increased flow velocities at these narrowings. Most major blood-flow-stopping events occur at large plaques, which, prior to their rupture, produced very little if any stenosis.
From clinical trials, 20% is the average stenosis at plaques that subsequently rupture with resulting complete artery closure. Most severe clinical events do not occur at plaques that produce high-grade stenosis. From clinical trials, only 14% of heart attacks occur from artery closure at plaques producing a 75% or greater stenosis prior to the vessel closing.[citation needed]
If the fibrous cap separating a soft atheroma from the bloodstream within the artery ruptures, tissue fragments are exposed and released. These tissue fragments are very clot-promoting, containing collagen and tissue factor; they activate platelets and activate the system of coagulation. The result is the formation of a thrombus (blood clot) overlying the atheroma, which obstructs blood flow acutely. With the obstruction of blood flow, downstream tissues are starved of oxygen and nutrients. If this is the myocardium (heart muscle), angina (cardiac chest pain) or myocardial infarction (heart attack) develops.
Diagnosis
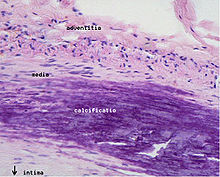 Microphotography of arterial wall with calcified (violet colour) atherosclerotic plaque (haematoxillin & eosin stain)
Microphotography of arterial wall with calcified (violet colour) atherosclerotic plaque (haematoxillin & eosin stain)Areas of severe narrowing, stenosis, detectable by angiography, and to a lesser extent "stress testing" have long been the focus of human diagnostic techniques for cardiovascular disease, in general. However, these methods focus on detecting only severe narrowing, not the underlying atherosclerosis disease. As demonstrated by human clinical studies, most severe events occur in locations with heavy plaque, yet little or no lumen narrowing present before debilitating events suddenly occur. Plaque rupture can lead to artery lumen occlusion within seconds to minutes, and potential permanent debility and sometimes sudden death.
Plaques that have ruptured are called complicated plaques. The extracellular matrix of the lesion breaks, usually at the shoulder of the fibrous cap that separates the lesion from the arterial lumen, exposing thrombogenic material, mainly collagen, and eventually causing thrombus formation. This thrombus will eventually grow and travel downstream until eventually occluding a narrow artery. Once the area is blocked, blood and oxygen will not be able to supply the vessels and will cause death of cells and lead to necrosis and poisoning. Serious complicated plaques can cause death of organ tissues, causing serious complications to that organ system.
Greater than 75% lumen stenosis used to be considered by cardiologists as the hallmark of clinically significant disease because it is typically only at this severity of narrowing of the larger heart arteries that recurring episodes of angina and detectable abnormalities by stress testing methods are seen. However, clinical trials have shown that only about 14% of clinically debilitating events occur at locations with this, or greater severity of stenosis. The majority of events occur due to atheroma plaque rupture at areas without narrowing sufficient enough to produce any angina or stress test abnormalities. Thus, since the later-1990s, greater attention is being focused on the "vulnerable plaque."[39]
Though any artery in the body can be involved, usually only severe narrowing or obstruction of some arteries, those that supply more critically important organs are recognized. Obstruction of arteries supplying the heart muscle result in a heart attack. Obstruction of arteries supplying the brain result in a stroke. These events are life-changing, and often result in irreversible loss of function because lost heart muscle and brain cells do not grow back to any significant extent, typically less than 2%.
Over the last couple of decades, methods other than angiography and stress-testing have been increasingly developed as ways to better detect atherosclerotic disease before it becomes symptomatic. These have included both (a) anatomic detection methods and (b) physiologic measurement methods.
Examples of anatomic methods include: (1) coronary calcium scoring by CT, (2) carotid IMT (intimal media thickness) measurement by ultrasound, and (3) intravascular ultrasound (IVUS).
Examples of physiologic methods include: (1) lipoprotein subclass analysis, (2) HbA1c, (3) hs-CRP, and (4) homocysteine.
The example of the metabolic syndrome combines both anatomic (abdominal girth) and physiologic (blood pressure, elevated blood glucose) methods.
Advantages of these two approaches: The anatomic methods directly measure some aspect of the actual atherosclerotic disease process itself, thus offer potential for earlier detection, including before symptoms start, disease staging and tracking of disease progression. The physiologic methods are often less expensive and safer and changing them for the better may slow disease progression, in some cases with marked improvement.
Disadvantages of these two approaches: The anatomic methods are generally more expensive and several are invasive, such as IVUS. The physiologic methods do not quantify the current state of the disease or directly track progression. For both, clinicians and third party payers have been slow to accept the usefulness of these newer approaches.
Treatment
If atherosclerosis leads to symptoms, some symptoms such as angina pectoris can be treated. Non-pharmaceutical means are usually the first method of treatment, such as cessation of smoking and practicing regular exercise. If these methods do not work, medicines are usually the next step in treating cardiovascular diseases, and, with improvements, have increasingly become the most effective method over the long term. Most medicines for atherosclerosis are patented, allowing manufacturers to enjoy higher prices than non-patented medicines; and they may cause unwanted side-effects.
Statins
In general, the group of medications referred to as statins has been the most popular and are widely prescribed for treating atherosclerosis. They have relatively few short-term or longer-term undesirable side-effects, and several clinical trials comparing statin treatment with placebo have fairly consistently shown strong effects in reducing atherosclerotic disease 'events' and generally ~25% comparative mortality reduction, although one study design, ALLHAT,[40] was less strongly favorable.
The newest statin, rosuvastatin, has been the first to demonstrate regression of atherosclerotic plaque within the coronary arteries by IVUS (intravascular ultrasound evaluation).[7] The study was set up to demonstrate effect primarily on atherosclerosis volume within a 2 year time-frame in people with active/symptomatic disease (angina frequency also declined markedly) but not global clinical outcomes, which was expected to require longer trial time periods; these longer trials remain in progress.
However, for most people, changing their physiologic behaviors[clarification needed], from the usual high risk to greatly reduced risk, requires a combination of several compounds, taken on a daily basis and indefinitely. More and more human treatment trials have been done and are ongoing that demonstrate improved outcome for those people using more-complex and effective treatment regimens that change physiologic behaviour patterns to more closely resemble those that humans exhibit in childhood at a time before fatty streaks begin forming.
The statins, and some other medications, have been shown to have antioxidant effects, possibly part of their basis for some of their therapeutic success[citation needed] in reducing cardiac 'events'.
The success of statin drugs in clinical trials is based on some reductions in mortality rates, however by trial design biased toward men and middle-age, the data is as, as yet, less strongly clear for women and people over the age of 70.[41] For example, in the Scandinavian Simvastatin Survival Study (4S), the first large placebo-controlled, randomized clinical trial of a statin in people with advanced disease who had already suffered a heart attack, the overall mortality rate reduction for those taking the statin, vs. placebo, was 30%. For the subgroup of people in the trial who had Diabetes Mellitus, the mortality rate reduction between statin and placebo was 54%. 4S was a 5.4-year trial that started in 1989 and was published in 1995 after completion. There were three more dead women at trial's end on statin than in the group on placebo; whether this was due to chance or some relation to the statin remains unclear. The ASTEROID trial has been the first to show actual disease volume regression[7] (see page 8 of the paper, which shows cross-sectional areas of the total heart artery wall at start and 2 years of rosuvastatin 40 mg/day treatment); however, its design was not able to "prove" the mortality reduction issue since it did not include a placebo group: the individuals offered treatment within the trial had advanced disease, and treatment with placebo was judged to be unethical.
Primary and secondary prevention
Combinations of statins, niacin, intestinal cholesterol absorption-inhibiting supplements (ezetimibe and others, and to a much lesser extent fibrates) have been the most successful in changing common but sub-optimal lipoprotein patterns and group outcomes. In the many secondary prevention and several primary prevention trials, several classes of lipoprotein-expression-altering (less correctly termed "cholesterol-lowering") agents have consistently reduced not only heart attack, stroke and hospitalization but also all-cause mortality rates. The first of the large secondary prevention comparative statin/placebo treatment trials was the Scandinavian Simvastatin Survival Study (4S)[42] with over fifteen more studies extending through to the more recent ASTEROID[43] trial published in 2006. The first primary prevention comparative treatment trial was AFCAPS/TexCAPS[44] with multiple later comparative statin/placebo treatment trials including EXCEL,[45] ASCOT[46] and SPARCL.[47][48] While the statin trials have all been clearly favorable for improved human outcomes, only ASTEROID showed evidence of atherosclerotic regression (slight). Both human and animal trials that showed evidence of disease regression used more aggressive combination agent treatment strategies, which nearly always included niacin.[14]
Diet and dietary supplements
Niacin (vitamin B3), in pharmacologic doses, (generally 1,000 to 3,000 mg/day, but starting with much lower doses increased over several weeks, to avoid side-effects[49]) tends to improve (a) HDL levels, size and function, (b) shift LDL particle distribution to larger particle size and (c) lower lipoprotein(a), an atherosclerosis promoting genetic variant of LDL. Additionally, individual responses to daily niacin, while mostly evident after a month at effective doses, tends to continue to slowly improve further over time. (However, careful patient understanding of how to achieve this without nuisance symptoms is needed, though not often achieved.) Research work on increasing HDL particle concentration and function, beyond the usual niacin effect/response, even more important, is slowly advancing. Niacin is supplied in many OTC and prescription formulations; non-prescription formulations recommend much lower doses as they are sold as nutritional supplements, not regulated medications.[49]
Dietary changes to achieve benefit have been more controversial, generally far less effective and less widely adhered to with success. One key reason for this is that most cholesterol, typically 80-90%, within the body is created and controlled by internal production by all cells in the body (true of all animals), with typically slightly greater relative production by hepatic/liver cells. (Cell structure relies on fat membranes to separate and organize intracellular water, proteins and nucleic acids and cholesterol is one of the components of all animal cell membranes.)
While the absolute production quantities vary with the individual, group averages for total human body content of cholesterol within the U.S. population commonly run about 35,000 mg (assuming lean build; varies with body weight and build) and about 1,000 mg/day ongoing production. Dietary intake plays a smaller role, 200–300 mg/day being common values; for pure vegetarians, essentially 0 mg/day, but this typically does not change the situation very much because internal production increases to largely compensate for the reduced intake. For many, especially those with greater than optimal body mass and increased glucose levels, reducing carbohydrate (especially simple forms) intake, not fats or cholesterol, is often more effective for improving lipoprotein expression patterns, weight and blood glucose values. For this reason, medical authorities much less frequently promote the low dietary fat concepts than was commonly the case prior to about year 2005. However, evidence has increased that processed, particularly industrial non-enzymatic hydrogenation produced trans fats, as opposed to the natural cis-configured fats, which living cells primarily produce, is a significant health hazard.
Dietary supplements of Omega-3 oils, especially those from the muscle of some deep salt water living fish species, also have clinical evidence of significant protective effects as confirmed by 6 double blind placebo controlled human clinical trials.[citation needed]
Less robust evidence shows that homocysteine and uric acid levels, including within the normal range, promote atherosclerosis and that lowering these levels is helpful.[citation needed]
In animals Vitamin C deficiency has been confirmed as an important role in development of hypercholesterolemia and atherosclerosis, but due to ethical reasons placebo-controlled human studies are impossible to do.[50] Vitamin C acts as an antioxidant in vessels and inhibits inflammatory process.[51] It has therapeutic properties on high blood pressure and its fluctuation,[52][53] and arterial stiffness in diabetes.[54] Vitamin C is also a natural regulator of cholesterol[55] and higher doses (over 150 mg/kg daily) may confer significant protection against atherosclerosis even in the situation of elevated cholesterol levels.[56][57]
The scale of vitamin C benefits on cardiovascular system led several authors to theorize that vitamin C deficiency is the primary cause of cardiovascular diseases.[58] The theory was unified by twice Nobel prize winner Linus Pauling, and Matthias Rath (Rath's promotion of vitamins instead of effective medicines for treatment of serious diseases has been very strongly criticised by many reputable authorities, as discussed in detail elsewhere). They point out that vitamin C is produced by all mammals except mankind and the great apes. This is due to a genetic deficiency that arose with the common ancestor of human and apes. To survive humans and apes must eat sufficient vitamin C each day. Without vitamin C humans develop scurvy. Vitamin C is an essential element in insuring that the vascular system is strong and flexible. Pauling and Rath suggest that a deficiency causes weakness in the arterial system and the body compensates by trying to stiffen up the artery walls using other common blood elements. This causes the effect known as atherosclerosis. They suggest that clinical manifestations of cardiovascular diseases are merely overshoot of body defense mechanisms that are involved in stabilisation of vascular wall after it is weakened by the vitamin C deficiency and the subsequent collagen degradation. They discuss several metabolic and genetic predispositions (our inability to produce vitamin C at all being the main one) and their pathomechanism.[59]
The Unified Theory of Human Cardiovascular Disease suggests that atherosclerosis may be reversed and cured,[59] but there has been no testing or trial of Pauling's vitamin C theory.
Trials on Vitamin E have been made, and have generally not found a beneficial effect. It has been suggested that there may be a beneficial effect for some patients at high risk for atherosclerosis. A review of trials suggested that the lack of evidence for a beneficial effect may have been due to various specified shortcomings in the trial methodologies, such as testing vitamin E without concurrent use of vitamin C.[60]
Menaquinone (Vitamin K2), but not phylloquinone (Vitamin K1), intake is associated with reduced risk of CHD mortality, all-cause mortality and severe aortic calcification.[61][62][63]
Excess iron may be involved in the development of atherosclerosis,[64][65] but one study found reducing body iron stores in patients with symptomatic peripheral artery disease through phlebotomy did not significantly decrease all-cause mortality or death plus nonfatal myocardial infarction and stroke.[66] Further studies may be warranted.
Changes in diet may help prevent the development of atherosclerosis. Researchers at the Agricultural Research Service have found that avenanthramides, chemical compounds found in oats, may help reduce the inflammation of the arterial wall, which contributes to the development of atherosclerosis. Avenanthramides have anti-inflammatory properties that are linked to activating proinflammatory cytokines. Cytokines are proteins that are released by the cell to protect and repair tissues. Researchers found that these compounds in oats have the ability to reduce inflammation and thereby help prevent atherosclerosis.[67][68]
Surgical intervention
Other physical treatments, helpful in the short term, include minimally invasive angioplasty procedures that may include stents to physically expand narrowed arteries[69] and major invasive surgery, such as bypass surgery, to create additional blood supply connections that go around the more severely narrowed areas.
Prophylaxis
Patients at risk for atherosclerosis-related diseases are increasingly being treated prophylactically with low-dose aspirin and a statin. The high incidence of cardiovascular disease led Wald and Law[70] to propose a Polypill, a once-daily pill containing these two types of drugs in addition to an ACE inhibitor, diuretic, beta blocker, and folic acid. They maintain that high uptake by the general population by such a Polypill would reduce cardiovascular mortality by 80%. It must be emphasized however that this is purely theoretical, as the Polypill has never been tested in a clinical trial.
Medical treatments often focus predominantly on the symptoms. However, over time, clinical trials have shown treatments that focus on decreasing the underlying atherosclerosis processes—as opposed to simply treating symptoms—more effective.
In summary, the key to the more effective approaches has been better understanding of the widespread and insidious nature of the disease and to combine multiple different treatment strategies, not rely on just one or a few approaches. In addition, for those approaches, such as lipoprotein transport behaviors, which have been shown to produce the most success, adopting more aggressive combination treatment strategies has generally produced better results, both before and especially after people are symptomatic.
Because many blood thinners, particularly warfarin and salicylates such as aspirin, thin the blood by interfering with Vitamin K, there is recent evidence that blood thinners that work by this mechanism can actually worsen arterial calcification in the long term even though they thin the blood in the short term.[71][72][73][74]
Prognosis
Lipoprotein imbalances, upper normal and especially elevated blood sugar, i.e., diabetes and high blood pressure are risk factors for atherosclerosis; homocysteine, stopping smoking, taking anticoagulants (anti-clotting agents), which target clotting factors, taking omega-3 oils from fatty fish or plant oils such as flax or canola oils, exercising and losing weight are the usual focus of treatments that have proven to be helpful in clinical trials. The target serum cholesterol level should ideally not exceed 4 mmol/L (160 mg/dL), and triglycerides should not exceed 2 mmol/L (180 mg/dL).
Evidence has increased that diabetics, despite not having clinically detectable atherosclerotic disease, have more severe debility from atherosclerotic events over time than even non-diabetics who have already suffered atherosclerotic events. Thus diabetes has been upgraded to be viewed as an advanced atherosclerotic disease equivalent[clarification needed].
Controversy
The belief that high fat and cholesterol consumption causes atherosclerosis has been questioned. Because fat and cholesterol are the substances of which plaque consists, they are both considered to be contributors to the cause of atherosclerosis, though this remains to be verified. Inflammation is considered to be a cause of atherosclerosis rather than fat and cholesterol.[75]
A team of scientists recently discovered the earliest known case of atherosclerosis in ancient Egyptian mummies. The findings could mean that some scientists may not understand heart disease as well as previously thought in regard to the conditions creating that condition. It may not be a modern disease at all and could have been common throughout human history.
This team began by running mummies through a CT scanner. "Our hypothesis was that they wouldn't have heart disease, because they were active, their diet was much different, they didn't have tobacco," he says. But they were wrong. One of the mummies the team scanned was a princess in her 40s. "That she would have atherosclerosis," the researcher says, "I think we're missing a risk factor. Right now we know that high blood pressure, smoking, cholesterol, inactivity and other things cause atherosclerosis, but I think that we're less complete than we think."[76]
Research
An indication of the role of HDL on atherosclerosis has been with the rare Apo-A1 Milano human genetic variant of this HDL protein. A small short-term trial using bacterial synthetized human Apo-A1 Milano HDL in people with unstable angina produced fairly dramatic reduction in measured coronary plaque volume in only 6 weeks vs. the usual increase in plaque volume in those randomized to placebo. The trial was published in JAMA in early 2006. Ongoing work starting in the 1990s may lead to human clinical trials—probably by about 2008. These may use synthesized Apo-A1 Milano HDL directly. Or they may use gene-transfer methods to pass the ability to synthesize the Apo-A1 Milano HDLipoprotein.
Methods to increase high-density lipoprotein (HDL) particle concentrations, which in some animal studies largely reverses and remove atheromas, are being developed and researched.
Niacin has HDL raising effects (by 10 - 30%) and showed clinical trial benefit in the Coronary Drug Project and is commonly used in combination with other lipoprotein agents to improve efficacy of changing lipoprotein for the better. However most individuals have nuisance symptoms with short term flushing reactions, especially initially, and so working with a physician with a history of successful experience with niacin implementation, careful selection of brand, dosing strategy, etc. are usually critical to success.
However, increasing HDL by any means is not necessarily helpful. For example, the drug torcetrapib is the most effective agent currently known for raising HDL (by up to 60%). However, in clinical trials it also raised deaths by 60%. All studies regarding this drug were halted in December 2006.[77] See CETP inhibitor for similar approaches.
The ERASE trial is a newer trial of an HDL booster, which has shown promise.[78]
The ASTEROID trial used a high-dose of rosuvastatin—the statin with typically the most potent dose/response correlation track record (both for LDLipoproteins and HDLipoproteins.) It found plaque (intima + media volume) reduction.[7] Several additional rosuvastatin treatment/placebo trials for evaluating other clinical outcomes are in progress.
The actions of macrophages drive atherosclerotic plaque progression. Immunomodulation of atherosclerosis is the term for techniques that modulate immune system function to suppress this macrophage action.[79] Immunomodulation has been pursued with considerable success in both mice and rabbits since about 2002. Plans for human trials, hoped for by about 2008, are in progress.
Research on genetic expression and control mechanisms is progressing. Topics include
- PPAR, known to be important in blood sugar and variants of lipoprotein production and function;
- The multiple variants of the proteins that form the lipoprotein transport particles.
Some controversial research has suggested a link between atherosclerosis and the presence of several different nanobacteria in the arteries, e.g., Chlamydophila pneumoniae, though trials of current antibiotic treatments known to be usually effective in suppressing growth or killing these bacteria have not been successful in improving outcomes.[80]
The immunomodulation approaches mentioned above, because they deal with innate responses of the host to promote atherosclerosis, have far greater prospects for success.
See also
- Angiogram
- Arterial stiffness
- Atheroma
- Chelation therapy
- Coronary circulation
- Coronary catheterization
- Fatty streaks
- Monckeberg's arteriosclerosis
- Intravascular ultrasound
Footnotes
- ^ Maton, Anthea; Roshan L. Jean Hopkins, Charles William McLaughlin, Susan Johnson, Maryanna Quon Warner, David LaHart, Jill D. Wright (1993). Human Biology and Health. Englewood Cliffs, New Jersey, USA: Prentice Hall. ISBN 0-13-981176-1. OCLC 32308337.
- ^ Ross R (April 1993). "The pathogenesis of atherosclerosis: a perspective for the 1990s". Nature 362 (6423): 801–9. doi:10.1038/362801a0. PMID 8479518.
- ^ Ross R; Ross, Russell (January 1999). "Atherosclerosis — An Inflammatory Disease". New England Journal of Medicine 340 (2): 115–26. doi:10.1056/NEJM199901143400207. PMID 9887164.
- ^ Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010 Jul;30(7):1282-92. Review. PubMed PMID 20554950.
- ^ Didangelos A, Simper D, Monaco C, Mayr M (May 2009). "Proteomics of acute coronary syndromes.". Current atherosclerosis reports 11 (3): 188–95. doi:10.1007/s11883-009-0030-x. PMID 19361350. http://www.springerlink.com/content/8qjl665766630k8p/fulltext.pdf.
- ^ a b Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ (May 1987). "Compensatory enlargement of human atherosclerotic coronary arteries". N. Engl. J. Med. 316 (22): 1371–5. doi:10.1056/NEJM198705283162204. PMID 3574413.
- ^ a b c d Nissen. "Effect of Very High-Intensity Statin Therapy on Regression of Coronary Atherosclerosis–The ASTEROID Trial". JAMA. http://jama.ama-assn.org/cgi/reprint/jama;295/13/1556.pdf?ijkey=Md42dlk7z9TzyL8&keytype=finite.
- ^ Williams, KJ; Tabas, I (1995 May). "The response-to-retention hypothesis of early atherogenesis.". Arteriosclerosis, thrombosis, and vascular biology 15 (5): 551–61. PMID 7749869.
- ^ Sparrow, CP; Olszewski, J (1993 Jul). "Cellular oxidation of low density lipoprotein is caused by thiol production in media containing transition metal ions.". Journal of lipid research 34 (7): 1219–28. PMID 8103788.
- ^ Fabricant CG, Fabricant J (1999). "Atherosclerosis induced by infection with Marek's disease herpesvirus in chickens". Am Heart J 138 (5 Pt 2): S465–8. doi:10.1016/S0002-8703(99)70276-0. PMID 10539849.
- ^ Hsu HY, Nicholson AC, Pomerantz KB, Kaner RJ, Hajjar DP (August 1995). "Altered cholesterol trafficking in herpesvirus-infected arterial cells. Evidence for viral protein kinase-mediated cholesterol accumulation". J Biol Chem 270 (33): 19630–7. doi:10.1074/jbc.270.33.19630. PMID 7642651. http://www.jbc.org/cgi/pmidlookup?view=long&pmid=7642651.
- ^ Cheng J, Ke Q, Jin Z, Wang H, Kocher O, Morgan JP, Zhang J, Crumpacker CS (May 2009). Früh, Klaus. ed. "Cytomegalovirus Infection Causes an Increase of Arterial Blood Pressure". PLoS Pathog 5 (5): e1000427. doi:10.1371/journal.ppat.1000427. PMC 2673691. PMID 19436702. http://www.plospathogens.org/article/info%3Adoi%2F10.1371%2Fjournal.ppat.1000427.
- ^ Rath M, Pauling L (1992). "A unified theory of human cardiovascular disease leading the way to the abolition of this disease as a cause for human mortality". Journal of Orthomolecular Medicine 7 (1): 5–15. http://orthomolecular.org/library/jom/1992/pdf/1992-v07n01-p005.pdf.
- ^ a b Blankenhorn DH, Hodis HN (August 1993). "Atherosclerosis--reversal with therapy". The Western journal of medicine 159 (2): 172–9. PMC 1022223. PMID 8212682. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1022223.
- ^ a b c d e f g h i j k l m n o p q r s Mitchell, Richard Sheppard; Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson (2007). Robbins Basic Pathology: With STUDENT CONSULT Online Access. Philadelphia: Saunders. pp. 345. ISBN 1-4160-2973-7. 8th edition.
- ^ Narain VS, Gupta N, Sethi R, Puri A, Dwivedi SK, Saran RK, Puri VK. Clinical correlation of multiple biomarkers for risk assessment in patients with acute coronary syndrome. Indian Heart J. 2008 Nov-Dec;60(6):536-42.
- ^ Rinehart, J. F., and Greenberg, L. D. 1949. Arteriosclerotic lesions in pyridoxine-deficient monkeys. Amer. J. Path. 25:481-491.
- ^ Rinehart, J. F., and Greenberg, L. D. 1956. Vitamin B6 deficiency in the Rhesus monkey with particular reference to the occurrence of atherosclerosis, dental caries, and hepatic cirrhosis. Amer. J. Clin. Nutr. 4:318-325.
- ^ Gruberg, E. R., Raymond, S. A. 1981. Beyond Cholesterol: Vitamin B6, Arteriosclerosis, and Your Heart. 1st ed. New York: St. Martin's Press.
- ^ Borissoff JI, Spronk HM, Heeneman S, ten Cate H. Is thrombin a key player in the 'coagulation-atherogenesis' maze? Cardiovasc Res. 2009;82(3):392-403. PMID19228706.http://cardiovascres.oxfordjournals.org/content/82/3/392.long
- ^ Borissoff JI, Heeneman S, Kilinc E, Kassak P, Van Oerle R, Winckers K, Govers-Riemslag JW, Hamulyak K, Hackeng TM, Daemen MJ, ten Cate H, Spronk HM. Early atherosclerosis exhibits an enhanced procoagulant state. Circulation. 2010;122(8):821-830. PMID20697022. http://circ.ahajournals.org/cgi/content/full/122/8/821
- ^ Borissoff JI, Spronk HM, ten Cate H. The hemostatic system as a modulator of atherosclerosis. N Engl J Med. 2011;364(18):1746-1760. PMID21542745. http://www.nejm.org/doi/full/10.1056/NEJMra1011670
- ^ Food and nutrition board, institute of medicine of the national academies (2005). Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids (Macronutrients). National Academies Press. pp. 481–484. http://www.nap.edu/openbook.php?record_id=10490&page=481.
- ^ Food and nutrition board, institute of medicine of the national academies (2005). Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids (Macronutrients). National Academies Press. pp. 494–505. http://www.nap.edu/openbook.php?record_id=10490&page=494.
- ^ Bhatt DL, Topol EJ (July 2002). "Need to test the arterial inflammation hypothesis". Circulation 106 (1): 136–40. doi:10.1161/01.CIR.0000021112.29409.A2. PMID 12093783. http://circ.ahajournals.org/cgi/content/full/circulationaha;106/1/136.
- ^ Griffin M, Frazer A, Johnson A, Collins P, Owens D, Tomkin GH (1998). "Cellular cholesterol synthesis--the relationship to post-prandial glucose and insulin following weight loss". Atherosclerosis. 138 (2): 313–8. doi:10.1016/S0021-9150(98)00036-7. PMID 9690914.
- ^ King, Cr; Knutson, Kl; Rathouz, Pj; Sidney, S; Liu, K; Lauderdale, Ds (December 2008). "Short sleep duration and incident coronary artery calcification". JAMA : the journal of the American Medical Association 300 (24): 2859–66. doi:10.1001/jama.2008.867. PMC 2661105. PMID 19109114. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2661105.
- ^ "Food Pyramids: Nutrition Source, Harvard School of Public Health". http://www.hsph.harvard.edu/nutritionsource/pyramids.html. Retrieved 2007-11-25.
- ^ Taubes G (March 2001). "Nutrition. The soft science of dietary fat". Science 291 (5513): 2536–45. doi:10.1126/science.291.5513.2536. PMID 11286266. http://www.sciencemag.org/cgi/pmidlookup?view=long&pmid=11286266.
- ^ Song JH, Fujimoto K, Miyazawa T (2000). "Polyunsaturated (n-3) fatty acids susceptible to peroxidation are increased in plasma and tissue lipids of rats fed docosahexaenoic acid-containing oils". J. Nutr. 130 (12): 3028–33. PMID 11110863.
- ^ Yap SC, Choo YM, Hew NF, et al. (1995). "Oxidative susceptibility of low density lipoprotein from rabbits fed atherogenic diets containing coconut, palm, or soybean oils". Lipids 30 (12): 1145–50. doi:10.1007/BF02536616. PMID 8614305.
- ^ Greco AV, Mingrone G (1990). "Serum and biliary lipid pattern in rabbits feeding a diet enriched with unsaturated fatty acids". Exp Pathol 40 (1): 19–33. PMID 2279534.
- ^ Mattes RD (2005). "Fat taste and lipid metabolism in humans". Physiol. Behav. 86 (5): 691–7. doi:10.1016/j.physbeh.2005.08.058. PMID 16249011. http://linkinghub.elsevier.com/retrieve/pii/S0031-9384(05)00397-5. "The rancid odor of an oxidized fat is readily detectable"
- ^ Dobarganes C, Márquez-Ruiz G (2003). "Oxidized fats in foods". Curr Opin Clin Nutr Metab Care 6 (2): 157–63. doi:10.1097/00075197-200303000-00004. PMID 12589185.
- ^ How Bad Are Cooking Oils? by Udo Erasmus, PhD
- ^ supplements, FDA. "Dietary Supplements". http://www.fda.gov/Food/DietarySupplements/default.htm.
- ^ Schwartz, CJ; Valente AJ, Sprague EA, Kelley JL, Cayatte AJ, Mowery J. (Dec 1992). "Atherosclerosis. Potential targets for stabilization and regression". Circulation 86 ((6 Suppl)): III117–123. PMID 1424045.
- ^ "Coronary atherosclerosis - the fibrous plaque with calcification". www.pathologyatlas.ro. http://www.pathologyatlas.ro/coronary-atherosclerosis-fibrous-plaque.php. Retrieved 2010-03-25.
- ^ Maseri A, Fuster V (2003). "Is there a vulnerable plaque?". Circulation 107 (16): 2068–71. doi:10.1161/01.CIR.0000070585.48035.D1. PMID 12719286.
- ^ The Allhat Officers And Coordinators For The Allhat Collaborative Research Group, (2002). "Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT-LLT)". JAMA 288 (23if the ti is): 2998–3007. doi:10.1001/jama.288.23.2998. PMID 12479764.
- ^ Vos E, Rose CP (November 2005). "Questioning the benefits of statins". CMAJ 173 (10): 1207; author reply 1210. doi:10.1503/cmaj.1050120. PMC 1277053. PMID 16275976. http://www.cmaj.ca/cgi/content/full/173/10/1207-a.
- ^ T. E. Strandberg, S. Lehto, K. Pyörälä, A. Kesäniemi, H. Oksa (1997-01-11). "Cholesterol lowering after participation in the Scandinavian Simvastatin Survival Study (4S) in Finland". European Heart Journal 18 (18(11)): 1725–1727;. PMID 9402446. http://eurheartj.oxfordjournals.org/cgi/content/abstract/18/11/1725. Retrieved 2007-11-18.
- ^ Nissen SE, Nicholls SJ, Sipahi I, et al. (2006). "Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial" (PDF). JAMA 295 (13): 1556–65. doi:10.1001/jama.295.13.jpc60002. PMID 16533939. http://jama.ama-assn.org/cgi/reprint/jama;295/13/1556.pdf?ijkey=Md42dlk7z9TzyL8&keytype=finite.
- ^ Downs JR, Clearfield M, Weis S, et al. (May 1998). "Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study". JAMA : the journal of the American Medical Association 279 (20): 1615–22. doi:10.1001/jama.279.20.1615. PMID 9613910. http://jama.ama-assn.org/cgi/pmidlookup?view=long&pmid=9613910.
- ^ Bradford RH, Shear CL, Chremos AN, et al. (1991). "Expanded Clinical Evaluation of Lovastatin (EXCEL) study results. I. Efficacy in modifying plasma lipoproteins and adverse event profile in 8245 patients with moderate hypercholesterolemia". Arch. Intern. Med. 151 (1): 43–9. doi:10.1001/archinte.151.1.43. PMID 1985608.
- ^ Sever PS, Poulter NR, Dahlöf B, et al. (2005). "Reduction in cardiovascular events with atorvastatin in 2,532 patients with type 2 diabetes: Anglo-Scandinavian Cardiac Outcomes Trial--lipid-lowering arm (ASCOT-LLA)". Diabetes Care 28 (5): 1151–7. doi:10.2337/diacare.28.5.1151. PMID 15855581.
- ^ Linda Brookes, MSc. "SPARCL: Stroke Prevention by Aggressive Reduction in Cholesterol Levels". Medscape. http://www.medscape.com/viewarticle/536377. Retrieved 2007-11-19.
- ^ Amarenco P, Bogousslavsky J, Callahan AS, et al. (2003). "Design and baseline characteristics of the stroke prevention by aggressive reduction in cholesterol levels (SPARCL) study". Cerebrovascular diseases 16 (4): 389–95. doi:10.1159/000072562. PMID 14584489. http://content.karger.com/ProdukteDB/produkte.asp?Aktion=ShowAbstract&ProduktNr=224153&ArtikelNr=72562.
- ^ a b Medtv: niacin dosage to b increased to 3,000mg/day over about 10 weeks to avoid side-effects
- ^ Ginter E (2007). "Chronic vitamin C deficiency increases the risk of cardiovascular diseases". Bratisl Lek Listy 108 (9): 417–21. PMID 18225482.
- ^ Böhm F, Settergren M, Pernow J (2007). "Vitamin C blocks vascular dysfunction and release of interleukin-6 induced by endothelin-1 in humans in vivo". Atherosclerosis 190 (2): 408–15. doi:10.1016/j.atherosclerosis.2006.02.018. PMID 16527283.
- ^ Sato K, Dohi Y, Kojima M, Miyagawa K, Takase H, Katada E, Suzuki S. (2006). "Effects of ascorbic acid on ambulatory blood pressure in elderly patients with refractory hypertension". Arzneimittelforschung 56 (7): 535–40. PMID 16927536.
- ^ Gladys Block, Christopher D Jensen, Edward P Norkus, Mark Hudes and Patricia B Crawford (2008). "Vitamin C in plasma is inversely related to blood pressure and change in blood pressure during the previous year in young Black and White women". Nutrition Journal 7 (35): 535–40. doi:10.1186/1475-2891-7-35. PMC 2621233. PMID 19091068. http://www.nutritionj.com/content/7/1/35.
- ^ Brian A. Mullan; Ian S. Young; Howard Fee; David R. McCance (2002). "Ascorbic Acid Reduces Blood Pressure and Arterial Stiffness in Type 2 Diabetes". Hypertension 40 (6): 804–9. doi:10.1161/01.HYP.0000039961.13718.00. PMID 12468561. http://hyper.ahajournals.org/cgi/content/abstract/40/6/804.
- ^ HJ Harwood Jr, YJ Greene and PW Stacpoole (June 5, 1986). "Inhibition of human leukocyte 3-hydroxy-3-methylglutaryl coenzyme A reductase activity by ascorbic acid. An effect mediated by the free radical monodehydroascorbate". J. Biol. Chem. 261 (16): 7127–7135. PMID 3711081. http://www.jbc.org/cgi/reprint/261/16/7127.
- ^ Das S, Ray R, Snehlata, Das N, Srivastava LM (2006). "Effect of ascorbic acid on prevention of hypercholesterolemia induced atherosclerosis". Mol Cell Biochem 285 (1–2): 143–7. doi:10.1007/s11010-005-9070-x. PMID 16479321.
- ^ Klenner F.R. (1974). "Significance of high daily intake of ascorbic acid in preventive medicine". J. Int. Acad. Prev. Med. 1: 45–49. http://www.seanet.com/%7Ealexs/ascorbate/197x/klenner-fr-j_int_assn_prev_med-1974-v1-n1-p45.htm.
- ^ Stone, I. (1972). The Healing Factor: Vitamin C Against Disease. Grosset and Dunlap, New York/Perigee Books, published by The Putnam Publishing Group. ISBN 0-399-50764-7. http://www.roccomanzi.it/IMP-VITAMINERALI/SCIENZIATI/scienziati-docu/stone/HEALINGFACTVitaC-TUTTO-ING_file/HEALINGFACTVitaC-TUTTO-ING.htm.
- ^ a b Rath M, Pauling L (1992). "A Unified Theory of Human Cardiovascular Disease Leading the Way to the Abolition of This Disease as a Cause for Human Mortality" (PDF). Journal of Orthomolecular Medicine 7 (1): 5–15. http://www.orthomolecular.org/library/jom/1992/pdf/1992-v07n01-p005.pdf.
- ^ Robinson I, de Serna DG, Gutierrez A, Schade DS (2006). "Vitamin E in humans: an explanation of clinical trial failure". Endocr Pract 12 (5): 576–82. PMID 17002935.
- ^ Geleijnse JM, Vermeer C, Grobbee DE, et al. (2004). "Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: the Rotterdam Study". J. Nutr. 134 (11): 3100–5. PMID 15514282.
- ^ Erkkilä AT, Booth SL (2008). "Vitamin K intake and atherosclerosis". Curr. Opin. Lipidol. 19 (1): 39–42. doi:10.1097/MOL.0b013e3282f1c57f. PMID 18196985.
- ^ Wallin R, Schurgers L, Wajih N (2008). "Effects of the blood coagulation vitamin K as an inhibitor of arterial calcification". Thromb. Res. 122 (3): 411–7. doi:10.1016/j.thromres.2007.12.005. PMC 2529147. PMID 18234293. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2529147.
- ^ Brewer GJ (2007). "Iron and copper toxicity in diseases of aging, particularly atherosclerosis and Alzheimer's disease". Exp. Biol. Med. (Maywood) 232 (2): 323–35. PMID 17259340.
- ^ Sullivan JL, Mascitelli L (2007). "[Current status of the iron hypothesis of cardiovascular diseases]" (in Italian). Recenti Prog Med 98 (7–8): 373–7. PMID 17685184.
- ^ Zacharski LR, Chow BK, Howes PS, et al. (2007). "Reduction of iron stores and cardiovascular outcomes in patients with peripheral arterial disease: a randomized controlled trial". JAMA 297 (6): 603–10. doi:10.1001/jama.297.6.603. PMID 17299195.
- ^ Agricultural Research Service-funded research by Mohsen Meydani
- ^ Nie, Wise, Peterson, and Meydani, Atherosclerosis journal: Avenanthramide, a polyphenol from oats, inhibits vascular smooth muscle cell proliferation and enhances nitric oxide production
- ^ "Heart disease and stents". Cypher Stent. http://www.cypherusa.com/cypher-j2ee/cypherjsp/main_splash/stent.jsp. Retrieved 2008-04-01.
- ^ Wald NJ, Law MR (June 2003). "A strategy to reduce cardiovascular disease by more than 80%". BMJ (Clinical research ed.) 326 (7404): 1419. doi:10.1136/bmj.326.7404.1419. PMC 162259. PMID 12829553. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=162259.
- ^ Price PA, Faus SA, Williamson MK (February 2000). "Warfarin-induced artery calcification is accelerated by growth and vitamin D". Arteriosclerosis, thrombosis, and vascular biology 20 (2): 317–27. PMID 10669626. http://atvb.ahajournals.org/cgi/pmidlookup?view=long&pmid=10669626.
- ^ Geleijnse JM, Vermeer C, Grobbee DE, et al. (November 2004). "Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: the Rotterdam Study". J Nutr. 134 (11): 3100–5. PMID 15514282. http://jn.nutrition.org/cgi/content/full/134/11/3100.
- ^ "Linus Pauling Institute at Oregon State University". lpi.oregonstate.edu. http://lpi.oregonstate.edu/infocenter/vitamins/vitaminK/. Retrieved 2010-03-25.
- ^ "GlycoDocs.org". glycodocs.org. http://www.glycodocs.org/wellness/index.php. Retrieved 2010-03-25.
- ^ http://circ.ahajournals.org/cgi/content/full/105/9/1135
- ^ http://www.npr.org/2011/04/09/135269340/egyptian-mummies-diagnosed-with-clogged-arteries?ft=1&f=1001 Retrieved April-10-11
- ^ Barter PJ, Caulfield M, Eriksson M, et al. (November 2007). "Effects of torcetrapib in patients at high risk for coronary events". N Engl J Med. 357 (21): 2109–22. doi:10.1056/NEJMoa0706628. PMID 17984165. http://content.nejm.org/cgi/content/full/NEJMoa0706628.
- ^ Sue Hughes (March 26, 2007). "ERASE: New HDL mimetic shows promise". HeartWire. http://www.theheart.org/article/779839.do.
- ^ Jan Nilsson; Göran K. Hansson; Prediman K. Shah (2005). "Immunomodulation of Atherosclerosis – Implications for Vaccine Development—ATVB In Focus". Arteriosclerosis, Thrombosis, and Vascular Biology 5 (1): 18–28. doi:10.1161/01.ATV.0000149142.42590.a2. PMID 15514204. http://atvb.ahajournals.org/cgi/content/abstract/atvbaha;25/1/18.
- ^ M Stitzinger (2007). "Lipids, inflammation and atherosclerosis" (pdf). The digital repository of Leiden University. https://openaccess.leidenuniv.nl/dspace/bitstream/1887/9729/11/01.pdf. Retrieved 2007-11-02. "Results of clinical trials investigating anti-chlamydial antibiotics as an addition to standard therapy in patients with coronary artery disease have been inconsistent. Therefore, Andraws et al. conducted a meta- analysis of these clinical trials and found that evidence available to date does not demonstrate an overall benefit of antibiotic therapy in reducing mortality or cardiovascular events in patients with coronary artery disease."
External links
- Atherosclerosis at the Open Directory Project
- A four-minute animation of the atherosclerosis process, entitled "Pathogenesis of Acute MI," commissioned by Paul M. Ridker, MD, MPH, FACC, FAHA, at the Harvard Medical School, can be viewed at pri-med.com.
Categories:- Diseases of arteries, arterioles and capillaries
- Vascular diseases




Wikimedia Foundation. 2010.